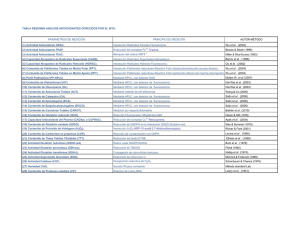
Incidencias y Mantenimiento en HPLC
Anuncio

Incidencias y Mantenimiento en HPLC Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 Incidencias y Mantenimiento en HPLC Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 -1- Incidencias y Mantenimiento en HPLC 1 INTRODUCCIÓN ................................................................................................................... - 4 - 2 EL CROMATÓGRAFO LÍQUIDO BÁSICO ....................................................................... - 6 2.1 VOLUMEN EXTRA EN COLUMNA(V0) .......................................................................................... - 9 2.2 CRITERIOS PARA UN MANTENIMIENTO LÓGICO .................................................................... - 11 2.3 REGLAS DE ORO EN EL DIAGNÓSTICO DE AVERÍAS ............................................................... - 12 2.4 PROBLEMAS TÍPICOS EN HPLC ............................................................................................. - 12 - 3 LA FASE MÓVIL .................................................................................................................. - 14 3.1 3.2 3.3 3.4 4 COMPATIBILIDAD DEL DISOLVENTE Y EQUILIBRADO DEL SISTEMA ...................................... - 14 PREPARACION DE LA FASE MOVIL.............................................................................................-19 DESGASIFICACIÓN DE LA FASE MÓVIL .................................................................................. - 33 FILTRACIÓN DE LA FASE MÓVIL ........................................................................................... - 40 - SISTEMAS DE BOMBEO .................................................................................................... - 46 4.1 4.2 4.3 4.4 4.5 4.6 4.7 FUNCIONES DE LA BOMBA .................................................................................................... - 46 BOMBA RECÍPROCA DE PISTÓN SIMPLE ................................................................................ - 47 LA BOMBA RECÍPROCA DE DOBLE PISTÓN ........................................................................... - 49 LA BOMBA BINARIA DE GRADIENTE ..................................................................................... - 50 LA BOMBA CUATERNARIA DE GRADIENTE ........................................................................... - 51 FORMACIÓN DE GRADIENTES Y TIEMPO MUERTO (“DWELL-TIME”) .................................... - 52 INCIDENCIAS DE LA BOMBA .................................................................................................. - 55 - TUTORIAL 1 ................................................................................................................................... - 61 5 INYECTORES ............................................................................................................................. - 65 5.1 5.2 5.3 COMPONENTES DEL SISTEMA DE INYECCIÓN ........................................................................ - 65 AUTOMUESTREADORES ......................................................................................................... - 71 INCIDENCIAS DEL AUTOMUESTREADOR ................................................................................ - 76 - 6 COLUMNAS ................................................................................................................................ - 86 6.1 LIMPIEZA, PURGA Y ALMACENAMIENTO DE LA COLUMNA ................................................... - 86 - 6.2 INCIDENCIAS DE LA COLUMNA........................................................................................................................-90- 7. PICOS CON COLA .................................................................................................................... - 96 8. PICOS CON FRENTE .............................................................................................................. - 101 TUTORIAL 2 ................................................................................................................................. - 102 9 DETECTORES .................................................................................................................... - 104 9.1 9.2 9.3 9.4 9.5 9.6 EL DETECTOR IDEAL ........................................................................................................... - 104 EL DETECTOR UV/VISIBLE ................................................................................................. - 107 INCIDENCIAS Y MANTENIMIENTO DEL DETECTOR UV/VISIBLE .......................................... - 109 PICOS NEGATIVOS............................................................................................................................................-114EL DETECTOR DE LIGHT SCATTERING EVAPORATIVO ......................................................... - 116 EL DETECTOR ESPECTROMÉTRICO DE MASAS ..................................................................... - 118 - TUTORIAL 3 ................................................................................................................................. - 125 10.1 LÍNEAS DE BASE ................................................................................................................ - 128 10.1.1 LÍNEAS DE BASE ERRÁTICAS ............................................................................................. - 128 10.1.2 LÍNEA DE BASE CÍCLICA (CORTA FRECUENCIA) ........................................................ - 130 10.1.3 LÍNEA DE BASE CÍCLICA (LARGA FRECUENCIA)........................................................ - 133 10.1.4 LINEAS DE BASES CON DERIVA ................................................................................. - 136 10.1.5 LÍNEAS DE BASE CON RUIDO ..................................................................................... - 138 10.2 FORMAS DE PICO .............................................................................................................. - 140 10.2.1 ENSANCHAMIENTO DE LOS PICOS .............................................................................. - 140 10.2.2 DESDOBLAMIENTO DE PICOS Y HOMBROS ................................................................. - 143 10.2.4 PICOS CON FRENTE .................................................................................................... - 151 10.2.5 EFECTO MEMORIA Y PICOS FANTASMA ..................................................................... - 152 10.2.6 PICOS NEGATIVOS ..................................................................................................... - 154 10.3 TIEMPO DE RETENCIÓN..................................................................................................... - 156 Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 -2- Incidencias y Mantenimiento en HPLC 10.3.1 10.3.2 10.3.3 10.4 10.5 TIEMPOS DE RETENCIÓN FLUCTUANTES .................................................................... - 156 DECRECIMIENTO DEL TIEMPO DE RETENCIÓN ........................................................... - 160 AUMENTO DEL TIEMPO DE RETENCIÓN ..................................................................... - 162 INCIDENCIAS CON LA PRESIÓN .................................................................................. - 162 SENSIBILIDAD............................................................................................................ - 166 - Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 -3- Incidencias y Mantenimiento en HPLC 1 Introducción El objetivo de esta sección es: • • Proporcionar una visión general mantenimiento en HPLC. Detallar el contenido del curso. del curso de incidencias y Contenidos del curso • El Cromatógrafo Líquido Básico • Criterios para un mantenimiento lógico • Incidencias y Mantenimiento Buenas prácticas de mantenimiento para; Fase Móvil Bomba Sistemas de Inyección Columnas Detectores • Diagnóstico desde una perspectiva sintomática Diagnósticos empleando el cromatograma parámetros de operación del sistema; y observando los Apariencia de la línea de base Cromatográfica. Apariencia de la forma de los picos Variación de los Tiempos de Retención. Alteraciones de la presión. Sensibilidad. Recuperación. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 -4- Incidencias y Mantenimiento en HPLC Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 -5- Incidencias y Mantenimiento en HPLC 2 El Cromatógrafo Líquido Básico El objetivo de esta sección está destinado a: • • • • Ilustrar y describir los fundamentos en cromatografía líquida. Describir los enfoques para resolver de forma lógica las incidencias en cromatografía líquida. Enumerar las "Reglas de Oro" en la resolución de problemas en HPLC. Describir los problemas típicos en HPLC. La figura anterior muestra los componentes modulares y el recorrido típico del flujo en un cromatógrafo líquido. Los módulos del sistema HPLC están conectados a través de tubos capilares fabricados en material polimérico, Polieter-eter-cetona (PEEK), o en acero inoxidable. Para eliminar volúmenes extra en columna que generen picos cromatográficos anchos, los módulos deben conectarse entre sí empleando la mínima longitud de tubo posible. Botellas de Disolvente Los disolventes que conforman la fase móvil están contenidos en botellas que generalmente se encuentran en la parte superior del sistema HPLC. Dicha disposición mejora su suministro, ya que se consigue un descenso natural de la fase líquida por efecto sifón bajo influencia de la gravedad. Las botellas de los disolventes deben estar cerradas herméticamente a fin de prevenir la pérdida de solventes orgánicos por evaporación, aunque siempre debe existir una pequeña abertura para evitar la formación de vacío Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 -6- Incidencias y Mantenimiento en HPLC que dificulte el cebado de la línea. En un análisis de larga duración, la pérdida de solventes orgánicos por evaporación en el espacio en cabeza disminuirá la fuerza efectiva de elución de la fase móvil, lo que provocará cambios en la retención del analito. Por tanto, debido a efectos por gradientes en la concentración y evaporación parcial de la fase móvil, se pueden producir cambios a lo largo del tiempo, que provocarán que las características de la fase móvil varíen de las inicialmente preparadas. Una buena práctica consiste en cambiar el disolvente usado por disolvente fresco, en lugar de extremar su consumo a largos periodos que tiempo hasta casi agotarlos. Se debe evitar el empleo de film de laboratorio (Parafilm, etc...) para cubrir las botellas de los disolventes. El material del film contiene ftalatos y polímeros, que pueden migrar por extracción con el solvente en el espacio en cabeza, incrementando el ruido en la línea de base del cromatograma. El Espectrómetro de Masas es especialmente sensible a este tipo de contaminantes que generan picos en la línea de base de un espectro de masas obtenido por LC-MS. La filtración de los disolventes antes de su uso, especialmente si estos contienen aditivos como sales tampón y/o reactivos de pares de iones, asegura la eliminación de material particulado que puede dañar los componentes del sistema. Además, es recomendable instalar en las líneas de disolvente un sistema de filtración que prevenga la entrada de partículas al sistema HPLC. Estos filtros están hechos a menudo de vidrio; cuando se deba eliminar contaminación se lavará primero con ácido nítrico concentrado (6M), después se hará un lavado con H2O, el siguiente lavado se hará con MeOH y después se hará pasar H2O de nuevo, antes de volver a reinstalar el sistema. Los filtros no deben sonicarse pues el vidrio sinterizado se podría romper y astillar, bloqueando los filtros y disminuyendo la eficacia del sistema. El reciclado de la fase móvil está limitado únicamente a las separaciones que trabajen en isocrático, y es más útil en los métodos empleados para análisis de rutina que contengan bajas concentraciones de muestra, como es el caso de laboratorios de Control de Calidad (CQ). Los componentes de la muestra se diluyen en la fase móvil de forma que se requieren largos periodos de tiempo para apreciar trazas significativas de los compuestos en el cromatograma. Generalmente esto se evidencia como un aumento en la Línea de Base y/o Fondo, lo que se traduce en pérdida de sensibilidad para el analito. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 -7- Incidencias y Mantenimiento en HPLC Desgasificador La eliminación de gases disueltos en la fase móvil ayuda a prevenir la formación de burbujas en el sistema HPLC. Burbujas de aire en la bomba pueden provocar pérdida de eficacia en el cebado de ésta, errores en la formación de gradientes de disolventes y ruido en la línea de base debido a fluctuaciones en la presión. Las columnas del HPLC pueden perder rendimiento si circulan burbujas de aire a través de estas debido a la formación de “canales” por presión, estos se saturarán rápidamente con moléculas de analito lo que hará perder capacidad de retención de la columna por perdida de superficie activa. Las burbujas de aire formadas por desgasificación en la celda del detector pueden contribuir al ruido de la línea de base, lo que reduce los límites de detección esperados y/o los límites de cuantificación. La figura anterior muestra los efectos por “canalización” de la columna en la retención del analito. Los disolventes pueden ser desgasificados fuera de línea antes de su uso, o como suele ser más frecuente, mediante incorporación de un desgasificador en línea que permite la desgasificación in situ de los disolventes. Bomba HPLC La bomba del HPLC, o sistema de suministro de disolvente, es la responsable de conseguir una composición exacta de la fase móvil a velocidades de flujo constantes y proporcionar la fuerza necesaria para el transporte de dicha fase a través del relleno de la columna. Lo ideal sería que la bomba no produjese impulsos impredecibles en la presión, por ello la mayoría de los sistemas acostumbran a estar dotados de un mecanismo integrado de amortiguación para la presión (Damper) que ayuda a corregir dicho efecto. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 -8- Incidencias y Mantenimiento en HPLC Automuestreador Es necesario un sistema automatizado que inyecte la muestra en cabeza de columna del HPLC, sin detener o distorsionar el flujo de la fase móvil. El muestreador automático debe minimizar el efecto memoria del analito entre inyecciones, e incrementar la productividad gracias a funciones adicionales tales como la capacidad de derivatizar la muestra antes de la columna, adición de soluciones de estándar interno, etc… Columna La separación cromatográfica de los componentes de la muestra tiene lugar a través de la interacción específica entre el analito y la fase estacionaria de la columna. Para largos periodos de tiempo entre usos, la columna del HPLC debe almacenarse en condiciones correctas. Las columnas no deben ser sometidas a excesivo estrés físico, así como a golpes o caídas, que pueden alterar el relleno de la fase estacionaria empobreciendo su rendimiento. Para minimizar variaciones en los tiempos de retención absolutos es habitual termostatizar la columna a fin de evitar los efectos fluctuantes de la temperatura ambiente. Detector Existe una gran variedad de detectores, aunque debido a su costo, robustez, límites de detección y facilidad de uso, el detector más utilizado es el de absorbancia UV. Los detectores UV están disponibles en una sola longitud de onda, múltiples longitudes de onda o en configuración de diodo array, que por su capacidad de analizar la de pureza de picos y de realizar búsquedas frente a bibliotecas de espectros UV ha llegado a ser muy popular. Para análisis que requieran alta sensibilidad y selectividad es preferible el uso de detectores de fluorescencia, electroquímicos o espectrómetros de masas. Los detectores de índice de refracción son solo aconsejables si el resto de detectores no son aplicables o si la concentración del analito es alta. 2.1 Volumen Extra en Columna(V0) Una consideración importante en el diseño de un sistema HPLC es el volumen extra en columna. El volumen extra en columna representa el volumen total de todos los componentes del sistema y conducciones capilares que no están directamente involucrados en el proceso de separación, desde el punto de inyección de la muestra hasta la detección. Este volumen extra debe ser minimizado a fin de evitar la formación de picos anchos y una disminución en la eficacia de la columna además de perder resolución cromatográfica. Por lo tanto; Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 -9- Incidencias y Mantenimiento en HPLC • • • Cada módulo debe ser conectado con la mínima longitud de tubo capilar y el mínimo diámetro interno posible. Los sistemas de inyección y los detectores deben ser diseñados para tener el volumen interno más pequeño posible. La columna debe tener un relleno lo más uniforme posible con la distribución de tamaños de partícula más estrecha permisible. Ancho de Pico provocado por un incremento de Volumen-Extra en Columna Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 10 - Incidencias y Mantenimiento en HPLC 2.2 Criterios para un Mantenimiento Lógico Hay dos enfoques que pueden ser utilizados para identificar problemas en cromatografía líquida: Perspectiva desde los Componentes Tras identificar el problema, cada componente que pueda contribuir a ello o ser la causa del fallo, se examina de nuevo. Cada componente examinado se limpia o se reemplaza según convenga y mediante el sistema de chequeo del HPLC en cada etapa se identifica si el problema ha sido corregido o no. Este enfoque en la resolución de problemas permite conseguir una familiarización del analista con el equipamiento del HPLC, aún así, puede perderse mucho tiempo en localizar y corregir componentes defectuosos. Este enfoque puede orientarse hacia la búsqueda de pistas visualmente como fugas o altas presiones y así poder localizar problemas en los componentes “físicos” del sistema. Perspectiva Sintomática Este enfoque en la identificación de problemas se refiere específicamente a los efectos que el analista puede ver. Generalmente incluye un examen visual de la línea de base o de la forma y/o apariencia de los picos cromatográficos a fin de encontrar indicios de problemas en la selectividad de la separación o en los componentes del sistema. Variaciones en los tiempos de retención, cambios en la presión del sistema, perdidas de sensibilidad o en las recuperaciones del analito, se pueden estudiar a través de ambas perspectivas. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 11 - Incidencias y Mantenimiento en HPLC 2.3 Reglas de Oro en el Diagnóstico de Averías La combinación de ambos diagnósticos, a partir de los componentes y a partir de los síntomas, nos permite formular una serie de “Reglas de Oro en el Diagnóstico de Averías” con un carácter empírico: 1. 2. 3. 4. 5. Hacer sólo un cambio por operación. Asegurarse de que el problema realmente existe. Confirmarlo como mínimo dos veces. Colocar correctamente de nuevo los componentes. Si no se encontró ningún fallo reinstalar correctamente las partes reemplazadas durante la sustitución de los componentes, Desechar los componentes defectuosos. No almacenar componentes que se sabe son defectuosos; desecharlos para eliminar toda posibilidad de utilización posterior. Anotar siempre los componentes reemplazados. Generar un registro de históricos de reparación y mantenimiento del sistema HPLC. Esto permite una acción realmente preventiva al realizar un mantenimiento programado de todo el sistema que elimina la necesidad de responder sólo ante posibles situaciones de fallo. Dicho registro, además, ayudará al ingeniero de servicio técnico cuando tenga que diagnosticar un problema en el sistema. 2.4 Problemas Típicos en HPLC Existen tres enfoques principales para localizar e identificar problemas en HPLC, incluyendo: 1. Examen de los parámetros operativos del sistema. Uno de los parámetros que más información aporta y que puede ser observado a través del monitor es la Presión del sistema. Según varíe la lectura de la presión se puede deducir que: • Es demasiado alta debido a un bloqueo en el sistema • Es demasiado baja debido a fugas en el sistema • Hay fluctuaciones. 2. Examen visual del sistema HPLC que especificará que componentes son susceptibles de fallo. Las fugas en las conexiones de accesorios, al final de la columna, en la celda del detector, en el automuestreador, en el cabezal de la bomba, etc… pueden ser debidas a sobrepresión del sistema y ser la causa de una pérdida de presión del mismo. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 12 - Incidencias y Mantenimiento en HPLC 3. Examen del Cromatograma resultante. Línea de Base • • • ¿La línea de base cromatográfica es plana, o fluctúa cíclicamente? ¿La línea de base cromatográfica muestra ruido en exceso? Examinando la línea de base se puede obtener gran cantidad de información del estado del sistema HPLC siendo de gran ayuda en el diagnóstico problemas. Picos • • • • ¿La forma de los picos cromatográficos es simétrica, o presentan colas y/o desdoblamientos apreciables? ¿Los Tiempos de Retención Absolutos de los picos cromatográficos varían entre inyecciones? ¿La sensibilidad frente al analito ha disminuido? Monitorizando los cambios en la forma de los picos cromatográficos y en los tiempos de retención se puede obtener una considerable información sobre el estado del sistema HPLC. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 13 - Incidencias y Mantenimiento en HPLC 3 La Fase Móvil El objetivo de esta sección es; • • • • • Explicar la importancia de asegurar la compatibilidad del disolvente. Describir un protocolo genérico para la preparación de las fases móviles. Explicar la importancia de la desgasificación de la fase móvil. Enumerar y describir los modos en los que la desgasificación de la fase móvil pueden realizarse. Explicar la importancia en la filtración de la fase móvil. 3.1 Compatibilidad del Disolvente y Equilibrado del Sistema Es importante tener en cuenta la miscibilidad de un disolvente con otro. Los disolventes que no son miscibles se particionarán en dos fases, dando como resultado una eliminación incompleta del disolvente original del sistema HPLC. Componentes del sistema como la válvula de proporción, las cámaras del pistón del cabezal de la bomba, los poros del relleno de la columna y la celda del detector continuarán liberando pequeñas gotas del disolvente anterior incluso después del proceso de purga, lo que hará que éstas migren a través del sistema creando perturbaciones en la línea de base cromatográfica, así como burbujas del disolvente que se comportarán de manera análoga a burbujas de aire que pasan a través de la celda del detector. El ruido en la línea de base afectará a la sensibilidad del analito, así como a los límites de cuantificación o detección que se verán incrementados dando como resultado: Ruido en la línea de base causado por la presencia de aire en la célula de flujo. . Una línea de base ruidosa afectará a la sensibilidad frente a los analitos, y se aumentarán los límites de detección y de cuantificación. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 14 - Incidencias y Mantenimiento en HPLC La tabla siguiente muestra la compatibilidad de disolventes. Se debe tener en cuenta la fuerza de los disolventes que están siendo usados: si hay grandes desajustes en la fuerza de los disolventes, las sales de las soluciones tampón o incluso las muestras podrían precipitar en el inyector o en la columna, provocando un incremento de contrapresión operativa en el sistema. La tabla muestra la miscibilidad de los disolventes más comunes en HPLC El estudio de compatibilidad de varios disolventes para HPLC muestra una gran variedad de solventes “universales”, como el tetrahidrofurano (THF) que es miscible con la mayoría del resto de disolventes. El disolvente de uso más frecuente en limpiezas es con diferencia el isopropanol (IPA) debido a su miscibilidad con el resto de solventes en todos los rangos de concentraciones. Ello hace del isopropanol una elección muy popular como solvente intermedio cuando hay cambio de eluyentes de fase reversa a fase normal y viceversa. Un cambio entre solventes que son inmiscibles, o una mala sustitución de la fase móvil previamente usada por sistema HPLC, hacen que la composición del eluyente en la columna no sea uniforme. Esto puede provocar variaciones en los Tiempos de Retención, especialmente durante las primeras inyecciones de muestras. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 15 - Incidencias y Mantenimiento en HPLC Fluctuaciones en los Tiempos de Retención debido a un pobre equilibrado de la fase móvil o al uso de solventes inmiscibles También se pueden observar derivas o ruido en la línea de base debido a variaciones en el detector entre una fase móvil nueva y la anterior: Ejemplo de una línea de base con deriva (figura superior) y una línea de base errática (figura inferior) que pueden verse en un sistema no equilibrado Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 16 - Incidencias y Mantenimiento en HPLC Cada vez que se cambie el disolvente es necesario hacer pasar por todo el sistema un volumen del solvente nuevo equivalente a 10 veces el volumen de la columna. Para calcular este volumen empíricamente hay que determinar antes el volumen vacío efectivo de la columna. Si las dimensiones de la columna fueran 150 x 4,6 mm de diámetro interno (DI), el volumen interno se puede calcular multiplicando el área por la longitud. Como regla general el volumen vacío de la columna será aproximadamente el 68% del total de esta, siendo el resto el ocupado por el material de relleno. Aplicando la siguiente ecuación y estando todas las dimensiones en milímetros, se obtiene el volumen vacío efectivo de la columna en microlitros; Volumen Vacío Columna = πr2 x L x 0.68 Volumen Vacío Columna = π (2.3)2 x 150 x 0.68 Volumen Vacío Columna ≈ 1,700µL (1.7mL) Por tanto, si el flujo fuera 1.0mL/min, serían necesarios aproximadamente 17 min para haber pasado 10 volúmenes de la columna en el cambio. Use siempre gradientes para pasar de un disolvente al siguiente. La siguiente secuencia de intercambio entre solventes es necesaria para realizar la conversión entre fase reversa y fase normal a fin de eliminar la posibilidad de inmiscibilidades entre los disolventes, o precipitaciones de la muestra o las sales tampón; • • • • • • Fase móvil Elimina de la columna todos los analitos retenidos. H2O Cualquier tampón usado debe ser reemplazado solo con agua, por ejemplo, en una separación isocrática con 50:50 MeOH:50mM de KH2PO4, entonces los 50mM de KH2PO4 deben ser intercambiados por H2O y el sistema deberá ser purgado adecuadamente. H2O Retirar la columna del sistema y purgar solo con H2O al 100% para eliminar todas las posibles trazas de sales tampón. Isopropanol Isopropanol:Hexano (u otros disolventes “no localizadores”(*)) 50:50 Elución en Fase Normal Nota: ¡El Acetonitrilo y el Hexano son INMISCIBLES! (*) que no tengan la capacidad de interactuar con grupos específicos de la fase estacionaria) Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 17 - Incidencias y Mantenimiento en HPLC Nota: Los métodos que usan reactivos formadores de pares de iones requieren normalmente tiempos largos de equilibrado. Suele ser necesario purgar con un mínimo de 20 volúmenes equivalentes al volumen de la columna. Protocolo genérico de Arranque para un sistema HPLC Para evitar los problemas asociados a un mal equilibrado del sistema se sugiere el protocolo de arranque descrito a continuación: 1. 2. 3. 4. 5. 6. 7. 8. Encender los módulos del equipo. Preparar los disolventes, incluyendo filtración y desgasificación. Cebar las líneas de los solventes y la bomba. Establecer la composición de la fase móvil y el flujo tal y como lo especifique el método. Purgar el sistema sin la columna durante 5 min, chequeando si hay fugas. Detener el flujo e instalar la columna. Purgar el sistema con la columna en su sitio durante un tiempo para dicho flujo que sea equivalente a mínimo 10 volúmenes de la columna, chequeando si hay fugas. Equilibrar el sistema durante 10 min, controlando contrapresiones en el sistema. Si procede, purgar el automuestreador y volver a equilibrar durante 10 min Inyectar la/s muestra/s de chequeo si el sistema está estabilizado Evaluar para confirmar si el sistema HPLC es “apto para la finalidad prevista” Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 18 - Incidencias y Mantenimiento en HPLC 3.2 Preparación de la Fase Móvil Influencia del pH El pH de la fase móvil influye directamente sobre el estado de ionización y en consecuencia sobre el tiempo de retención del analito. El pH y la capacidad tamponadora de cualquier fase móvil requieren de una medición y una preparación exacta respectivamente, de lo contrario la selectividad en la separación variará dando como resultado tiempos de retención irreproducibles y formas de pico variables. El cromatograma siguiente muestra la separación de seis analitos de ácido débil en una fase móvil de pH 5.10. A ese pH los seis componentes quedan resueltos adecuadamente, con una resolución de 1.50 para el par de picos críticos 1 y 2 – separación mínima de pico recomendada en análisis cuantitativos Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 19 - Incidencias y Mantenimiento en HPLC Sin embargo, si el pH de la fase móvil se ajusta incorrectamente, o el error de medida en la calibración del pH es del orden de 0.10 unidades, la separación cromatográfica de los seis analitos anteriores presentaría el siguiente cromatograma; A un pH de 5.20 los seis analitos no se resuelven correctamente, con una resolución de tan solo 0.58 en la separación de los picos 5 y 6. Dicho valor es considerablemente menor a 1.50 y por consiguiente el área de cada pico no puede ser integrada correctamente para la cuantificación. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 20 - Incidencias y Mantenimiento en HPLC Cuando se cromatografían compuestos ionizables es una ventaja mantener los compuestos en su forma no ionizada. Los compuestos ionizados representan analitos altamente polares, que en condiciones de fase reversa eluyen rápidamente de la columna, lo que reduce las posibilidades de separarlos del resto de analitos ionizados. Usando soluciones tampón y ajustando el pH para forzar que los compuestos estén en forma no ionizada, los ácidos y bases débiles pueden separarse eficientemente en condiciones de fase reversa. 10 9 8 O R C O H 7 Weakly Basic 6 5 k’ 4 Weakly Acidic 3 2 1 0 2 pH = pK 6 4 O R C O 8 pH El pKa de una molécula puede usarse para determinar a que pH debe tamponarse la fase móvil para asegurar una buena reproducibilidad en los tiempos de retención: • • Para ácidos orgánicos el pH se ajusta 2 unidades por debajo de su pKa Para bases orgánicas el pH se ajusta 2 unidades por encima de su pKa. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 21 - Incidencias y Mantenimiento en HPLC Fuerza Tamponadora El cromatograma siguiente muestra el efecto de un cambio en la fuerza tamponadora durante una separación isocrática de seis aditivos alimentarios; ácido ascórbico, sorbato potásico, benzoato sódico, vainillina, cumarina y etil-vainillina, picos del 1 al 6 respectivamente. El primer cromatograma muestra la separación de seis analitos mediante un tampón de concentración 60mM. La pareja de picos 4 y 5 se separa con una resolución de 1.65, separación suficiente para poder cuantificarlos. El segundo cromatograma muestra la separación de los mismos seis analitos con un tampón de concentración 57 mM. Con tan solo una ligera reducción en la concentración del tampón, la resolución en la separación de la pareja de picos 4 y 5 pasa a ser de solo 0.97, separación insuficiente para una correcta cuantificación. Un cambio de 3mM en la concentración del tampón tiene efectos notables en la selectividad del sistema de separación. El resultado es una pareja de picos inadecuadamente separados y la imposibilidad de su análisis cuantitativo lo que nos demuestra la importancia de una pesada exacta. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 22 - Incidencias y Mantenimiento en HPLC Determinación del Peso y Pesada Cualquier pesada estará sujeta a errores que deben ser minimizados para asegurar la menor desviación posible en la cuantificación final obtenida. Se conseguirán mínimos errores siguiendo los siguientes criterios: Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 23 - Incidencias y Mantenimiento en HPLC Uso del equipo de medición adecuado: • El equipo de medición funciona correctamente, es decir, “es adecuado para el fin previsto” y está calibrado correctamente de manera que la fiabilidad de la medida obtenida es alta. • El procedimiento de medida y de registro utilizado es correcto. El “Límite Funcional de Exactitud” (FAL, del inglés “Functional Accuracy Limit”) de cualquier balanza utilizada en el pesaje de sales para la solución tampón u otros aditivos, está aceptado generalmente como un 0.1%, siendo este el % de error permitido en la medida. Es necesario determinar de antemano que tipo de lectura tendrá la balanza que va a ser utilizada, y por tanto “La Cantidad Mínima Permisible” que puede ser pesada (MAQ, del inglés “Minimum Allowable Quantity”). La MAQ para una balanza puede ser determinada del siguiente modo: MAQ = 100% / FAL (%) x lectura de la balanza. Es decir, para una balanza de cuatro cifras (de lectura 0.0001g) se tendrá una MAQ igual a: MAQ = 100% / 0.1% x 0.0001 = 0.1g (100mg) Por lo tanto, para determinaciones de peso con precisión para el ejemplo anterior, el límite inferior serían 100mg para una balanza de cuatro dígitos. Balanza de 6 decimales MAQ (FAL = 0.1%) = 1mg Balanza de 4 decimales MAQ (FAL = 0.1%) = 100mg Balanza de 3 decimales MAQ (FAL = 0.1%) = 1g Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 24 - Incidencias y Mantenimiento en HPLC Las tolerancias típicas en el pesaje para los tres modelos de balanzas descritos son: Tolerancias Típicas en el Pesaje para los modelos de Balanzas descritos Modelo de Balanza Plato de Carga Superior Semi-micro analítica Micro analítica Peso Máximo Tolerancia (g) (mg) 3,200 ±10 5,200 ±10 8,200 ±100 60 ±0.015 230 ±0.025 2.1 ±0.00025 5.1 ±0.0010 21 ±0.0020 Los sólidos ya pesados deben ser transferidos al vidrio adecuado - matraz aforado o probeta - y ser disueltos en un agitador magnético y/o ultrasonidos sin llegar a enrasar. Se deben evitar calentamientos pues pueden afectar la composición del disolvente. Elección del Recipiente para el Pesaje • • Es muy importante que el recipiente usado para pesar sea el apropiado a la muestra o material que están siendo pesados, es decir, sería inapropiado pesar 0.01g en un recipiente de 1Litro. Utilizar siempre vidrios limpios y secos – esto es especialmente importante para los vidrios de reloj, donde las contaminaciones del material y la adsorción de la muestra por el vidrio pueden causar errores. Fuentes de Deriva La precisión en la medida del peso con la balanza analítica puede sufrir deriva. Las fuentes de deriva incluyen: • • • • • Avería en el plato de la balanza analítica o en su sensor. Cambios en los sensores mecánicos o de presión de la balanza debidos a averías y cambios en la temperatura. Exposición del mecanismo de la balanza a un exceso de carga estática Cambios en el calibrado interno del aparato. Cambios en la presión y temperatura ambiente. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 25 - Incidencias y Mantenimiento en HPLC Debido a esta inherente susceptibilidad a la deriva es necesario calibrar regularmente las balanzas. Efectos Estáticos El mecanismo por el que la balanza percibe el peso mediante sensores de presión y sus sistemas electrónicos internos, pueden alterarse sustancialmente por la electricidad estática de los envases o recipientes, así como la carga de las muestras o las sustancias que están siendo pesadas. Los contenedores de vidrio ó plástico son particularmente susceptibles a este efecto ya que la densidad de la carga no se elimina fácilmente debido a su baja conductividad. El látex y los guantes de plástico son también conocidos transmisores de carga electrostática al plato de las balanzas. Las balanzas afectadas por electricidad estática presentan generalmente problemas de lecturas muy inestables o derivas continuas. Hay varias recomendaciones a seguir para contrarrestar este efecto: • • • • Se pueden adquirir balanzas con plato recubierto anti-estáticamente. Se puede poner un vaso de agua en el compartimento de la balanza para aumentar la humedad ambiente (Nota: Este método NO es recomendable si el aumento de la humedad afecta a la muestra por ser higroscópica (ver sección siguiente). Se puede adquirir una “pistola ionizante” y colocarla cerca del compartimento de la balanza a fin de neutralizar mediante iones con carga opuesta. Manipular los envases con pinzas o tenazas – usar solo los guantes de látex cuando se manipulen muestras tóxicas. Muestras Higroscópicas • • • • • Algunas muestras son particularmente susceptibles atraer o absorber humedad. El acetato de amonio, un tampón muy usado en LC-MS, es extremadamente higroscópico. Un síntoma de muestras higroscópicas es un constante aumento del peso debido a la absorción de humedad. Cualquier muestra que se sepa que es higroscópica puede pesarse en una balanza usando un vaso de precipitados con un tamiz molecular para reducir la humedad presente o procurando minimizar su exposición ambiental. Las huellas dactilares en los envases de las muestras pueden favorecer la adsorción de la humedad. Por ello es importante que los envases o los recipientes no se manipulen directamente, sino que se usen espátulas o pinzas para manipularlos. Si las muestras o los recipientes requieren enfriamiento previo para ser pesados, estos deben ser enfriados en un desecador para evitar la absorción de humedad. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 26 - Incidencias y Mantenimiento en HPLC Muestras Volátiles • • • Esta situación es la opuesta a tener muestras higroscópicas. Las muestras volátiles tienden a perder peso, obteniéndose una disminución constante en la lectura del peso. Muchos tampones y aditivos usados en LC-MS son volátiles, dicha volatilidad ayuda a prevenir contaminación o suciedad en la fuente de iones. Es posible pesar muestras en recipientes cerrados con tapa, si esta se manipula con precaución. Cualquier líquido que condense en la tapa puede crear alteraciones en pesadas sucesivas. Se deben usar siempre recipientes tapados cuando se pesen líquidos. Para todos los pesajes es mejor minimizar el área superficial de la muestra que está siendo pesada. Este enfoque minimiza la exposición atmosférica de la muestra, reduciendo la volatilización potencial. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 27 - Incidencias y Mantenimiento en HPLC Efectos de la Temperatura • Como regla general, la muestra a pesar debe acondicionarse a la temperatura de la balanza. Es el único modo de evitar errores por ascensión del aire y desviaciones causadas por corrientes en la superficie de la muestra. Dado que estos efectos aumentan proporcionalmente al volumen y a la superficie de la muestra, se debe asegurar que el tamaño del recipiente seleccionado esté en proporción adecuada al tamaño de la muestra a pesar, ello ayudará a minimizar dicho efecto. En todos los casos las medidas fundamentales son: • • • • Obtener tanta información como sea posible de la muestra a pesar. Evitar potenciales efectos electrostáticos mencionados anteriormente. Equilibrar la temperatura del envase y la muestra con el ambiente de la balanza. Minimizar el área superficial de la muestra expuesta a la atmósfera. El pH Metro y el Ajuste de pH El pH es uno de los parámetros más comúnmente determinados en soluciones tanto acuosas como no acuosas. Variaciones en el pH pueden provocar cambios significativos en la velocidad de reacción así como cambios en la solubilidad y biodisponibilidad de muchos principios activos con fines farmacéuticos. Como se ha descrito anteriormente, las características en la retención de muchas especies de analitos pueden cambiar por variaciones en el pH de la fase móvil y por tanto contribuir a formas de picos anormales, es decir, desdoblamientos y colas. El pH es un parámetro que indica la acidez o alcalinidad de una solución, se mide en una escala de 0 a 14, donde un valor de pH igual a 0 es muy “ácido” y un valor de 14 es muy “básico”. El pH mide la concentración, o más exactamente la actividad del Ion oxonio – H3O+, aunque en la solución el Ion oxonio está representado como un protón o Ion hidrógeno, H+. Generalmente se ajusta primero el pH del disolvente de la fase móvil antes de añadir el volumen de disolvente orgánico necesario. Este enfoque sistemático asegura exactitud en la concentración del Ion oxonio. Para asegurar homogeneidad mientras se adiciona el reactivo en el ajuste del pH, la solución puede ser agitada en una placa magnética, aunque se debe evitar calentamientos que puedan afectar la composición del disolvente. El reactivo para ajustar el pH puede añadirse gota a gota usando material plástico, vidrio o pipetas dispensadoras automáticas. El empleo de vidrio propicia la introducción de iones metálicos. Los iones Al3+ y Fe2+ inducirán actividad en los grupos silanol residuales de la columna analítica. La consecuencia de esto es la observación de colas en los picos Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 28 - Incidencias y Mantenimiento en HPLC cromatográficos, especialmente para aquellas especies de analitos que tienen grupos funcionales básicos. Los iones aducto Na+ y K+ pueden predominar en LC-MS, llegando a ser los picos base y más abundantes incluso que el ion protonado pseudo-molecular. Esto puede influir de manera considerable en la cuantificación. Determinación del pH El sistema de medición del pH consta de tres componentes: • • • Un electrodo de pH. Un electrodo de referencia. Un pH metro de alta impedancia. En la mayoría de los sistemas para medir el pH los electrodos de pH y de referencia están montados en el mismo dispositivo, conocido como “electrodo combinado”. Electrodo de Referencia El electrodo de referencia consiste en un cable de plata recubierto en cloruro de plata, una solución de relleno y una junta permeable (generalmente fritas de cerámica o membranas), a través del cual la solución de relleno migra lentamente dentro de la muestra que está siendo medida. En algunas partes de la frita apenas hay flujo neto de la solución de relleno, dicha región se llama junta de difusión. Electrodo de pH El electrodo de pH está hecho de un vidrio de composición especial, que es sensible a la concentración de iones hidrógeno. Dicho vidrio está compuesto generalmente por un metal alcalino que sostiene una reacción de intercambio de iones con los Iones hidrógeno de la solución test para generar una diferencia de potencial. Electrodos de pH Combinados Los electrodos combinados contienen los dos electrodos descritos anteriormente en un único cuerpo de vidrio. El potencial generado en la zona de unión se debe a los iones libres de hidrógeno presentes en la muestra. El potencial de referencia es generado por un elemento interno de Ag/AgCl que está en contacto con la solución relleno de referencia. El electrodo de medida emite un voltaje variable y el electrodo de referencia un voltaje constante para el medidor. El sistema funciona de manera análoga a la medida del voltaje de una batería por un voltímetro. El cable interior del electrodo de pH tiene la misma finalidad que el primer cable que va desde la batería hasta el Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 29 - Incidencias y Mantenimiento en HPLC voltímetro. El electrodo de referencia equivale al segundo cable que completa el circuito y permite medir el voltaje. La solución de relleno fluye del electrodo de referencia hasta la solución test, completándose el “circuito” con el electrodo de pH. Electrodo de pH Combinado ROSS® Agujero de Relleno: Solución relleno de KCl Electrodo de referencia Ag/AgCl Unión de frita cerámica referencia Bulbo detector de pH Para que el electrodo de pH combinado trabaje correctamente es importante que el bulbo detector se guarde limpio y sin restos de tampones, de lo contrario los iones libres que fluyan a través del bulbo serán inhibidos y por tanto las medidas de pH serán incorrectas. Debe asegurarse que la solución relleno de KCl está en el nivel correcto y que el agujero del relleno no está tapado mientras se mide el pH. El pH-metro El electrodo de pH tiene una resistencia interna muy alta, haciendo muy difícil la medida de los pocos milivoltios de salida que provocan los cambios de pH. Un pH-metro es básicamente un amplificador de alta impedancia que mide con precisión los diminutos cambios de voltaje del electrodo, mostrando el resultado directamente en unidades de pH o en milivoltios. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 30 - Incidencias y Mantenimiento en HPLC Soluciones Tampón y Características del Electrodo de pH Soluciones Tampón Los tampones son soluciones que mantienen un valor constante de pH con capacidad de resistir a los cambios de éste. Se utilizan para calibrar el electrodo de pH y el pH-metro. Se emplean para compensar las pequeñas diferencias que se observan entre la señal de producida por diferentes electrodos, así como los cambios producidos en los electrodos con el paso del tiempo. En consecuencia, el sistema debe ser calibrado periódicamente. Los tampones disponen de un amplio rango de valores del pH, viniendo estos en forma líquida premezclada o como polvo seco encapsulado según sea necesaria su preparación. Generalmente una solución tampón específica calibrará con una tolerancia menor a ±0.02 unidades de pH medido a 20ºC. La mayoría de los pH-metros requieren calibraciones a varios valores específicos de pH. Generalmente se realiza una calibración cerca del punto isopotencial – la señal emitida por un electrodo a pH 7 y a 25ºC son 0mV – y típicamente se hace una segunda medida a pH 4 o a pH 10. Es bueno elegir un tampón con un pH tan próximo como sea posible al valor del pH de la muestra a determinar, pues se asegura la mayor exactitud posible. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 31 - Incidencias y Mantenimiento en HPLC Adición del Disolvente Cuando se prepare una fase móvil mezcla de disolventes acuosos y orgánicos, el procedimiento correcto será adicionar el volumen de solvente orgánico necesario al volumen de solvente acuoso necesario. Si el volumen necesario de la fase móvil se prepara simplemente por adición de la fase acuosa con el solvente orgánico o viceversa, entonces los errores en la composición pueden deberse a contracción o expansión del volumen de la mezcla respecto a los volúmenes individuales de los disolventes. Este efecto se traducirá en cambios crecientes en los tiempos de retención de la muestra y en los factores de capacidad. La tabla siguiente muestra el efecto anterior sobre las características de retención del tolueno. Muestra que hay una variación significativa en el tiempo de retención del tolueno entre una fase móvil 60:40 MeOH:H2O preparada por adición de cada disolvente según haya: • • Suficiente MeOH para 40mL de H2O o Suficiente H2O para 60mL de MeOH en comparación al procedimiento correcto de adición de 60mL MeOH a 40mL H2O. En el procedimiento correcto se obtiene una fase móvil de composición y fuerza de elución intermedia, prueba de ello es el cambio en el tiempo de retención y el factor de capacidad del tolueno al preparar la fase móvil de este modo y los otros dos anteriores. Resultado según la Técnica de Preparación de la Fase Móvil sobre la Retención del Tolueno Fracción MeOH Volumétrica del Tiempo de Retención Factor de Capacidad (min) k' 60 5.40 2.72 62* 4.82 2.32 58** 5.87 3.05 (%) * 40mL de H2O llevados hasta 100mL con 60mL MeOH, Calculado el k' ** 60mL de MeOH llevados hasta 100mL con 40mL H2O, Calculado el k' Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 32 - Incidencias y Mantenimiento en HPLC 3.3 Desgasificación de la Fase Móvil La eliminación de gases, o desgasificación, es un fenómeno al que se someten todos los disolventes tanto orgánicos como acuosos usados en cromatografía. Cuando el HPLC está sometido a altas presiones la solubilidad de los gases atmosféricos O2 y N2 aumenta. Cuando los disolventes experimentan una brusca caída de presión, generalmente durante la mezcla en una válvula de proporción cuaternaria, un mezclador estático de un sistema binario, o al entrar en la celda del detector, los gases disueltos se desprenden de la solución formando burbujas. La generación de dichas burbujas puede ser perjudicial tanto para la columna como para el proceso cromatográfico. Los sistemas de bombeo más modernos están centrados en el diseño de doble pistón. Las burbujas de aire pueden retenerse en las válvulas antirretorno o en las cámaras de los pistones del cabezal de la bomba. Se observará una línea de base sinusoidal como resultado de aire retenido en el cabezal de la bomba: Línea de Base Sinusoidal generada por aire retenido en el cabezal de la bomba. En estos casos es importante eliminar las burbujas de aire antes de que estas pasen a la columna. La bomba debe purgarse inmediatamente y el flujo incrementarse a 5ml/min para eliminar el aire del cabezal de la bomba. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 33 - Incidencias y Mantenimiento en HPLC Una burbuja que entra en la columna forzará su recorrido a través del lecho de relleno de la fase estacionaria, provocando la formación de “canales”. Dicha canalización disminuye rápidamente el rendimiento de la columna ya que los analitos son capaces de atravesar la columna por dichos canales, lo que se traduce en un menor tiempo de retención sobre la fase estacionaria, saturándose rápidamente dichos huecos por moléculas de analito. Tal efecto conduce generalmente a la formación de frentes en la forma de los picos cromatográficos. Frentes y picos anchos observados en una columna dañada Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 34 - Incidencias y Mantenimiento en HPLC El paso de burbujas de gas al HPLC afectará a la línea de base cromatográfica generando ruido que se traduce en la presencia de picos fantasma. Además, si una burbuja de gas queda atrapada en la celda del detector, se generará ruido de forma aleatoria en forma de picos en el registro de la línea de base. El efecto neto será una disminución en la relación señal-ruido (S/N), con su correspondiente aumento en los límites de detección (LoD) y los límites de cuantificación (LoQ) del método. En cuanto la fase móvil entra en la celda de flujo del detector se produce una caída en la presión. Como resultado de esta caída en la presión, una pequeña cantidad del aire retenido en la fase móvil puede liberarse en la celda del detector. Dicha liberación de gas se traduce en un incremento del ruido en la línea de base: Incremento del ruido en la línea de base (figura superior) y “Spikes” (figura inferior) causados por burbujas de aire retenido y liberado en la celda del detector Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 35 - Incidencias y Mantenimiento en HPLC Es una práctica muy común ajustar la contrapresión mediante reguladores a la salida de la celda del detector a fin de mantener valores altos en la presión que minimicen la desgasificación hasta que el flujo de la fase móvil está suficientemente a distancia para prevenir el atrapamiento de las burbujas de aire formadas. Si el regulador de la presión no es un accesorio integrado del detector, suele ser suficiente la conexión de un tubo PEEK de 50cm de longitud y 0.17mm de diámetro interno antes del tubo de salida de desechos. Además, una pobre desgasificación de la fase móvil se traducirá en un mal cebado de la bomba y una pobre reproducibilidad de gradientes, lo que llevará a variaciones en los tiempos de retención del analito. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 36 - Incidencias y Mantenimiento en HPLC Métodos de Desgasificación Hay cuatro modos aceptados para desgasificar la fase móvil: • • • • Borboteo por Helio Desgasificación por Vacío Ultrasonidos Reflujo El filtrado de la fase móvil a vacío es el procedimiento que se emplea generalmente cuando se elimina materia particulada de una solución antes de su uso, dicho proceso desgasifica la fase móvil de manera indirecta. Aunque es una medida necesaria, especialmente cuando se usan soluciones tampón o fases móviles que contienen reactivos de par de iónico, se debe tener cuidado en evitar pérdidas selectivas de cualquier solvente orgánico que pueda conducir a variaciones en el tiempo de retención del analito. Borboteo por Helio El Borboteo por Helio elimina aproximadamente el 80% de los gases disueltos. El mecanismo de borboteo puede construirse simplemente conectando a un cilindro de helio un tubo de PTFE con una frita al final. Hay que sumergir la frita dentro de la fase móvil y ajustar el regulador para liberar una suave corriente de burbujas. En general, un litro de fase móvil se desgasificará por completo con un litro de helio. Una excesiva desgasificación con helio puede provocar la pérdida de la mayoría de los componentes volátiles de la fase móvil, generando variaciones en los tiempos de retención. La principal finalidad del borboteo por helio es disolver el N2 y el O2 que se extraen de la fase móvil gracias a su elevada solubilidad en helio. El helio tiene una solubilidad muy baja en la mayoría de las fases lo que permite la disolución de N2 y O2 en el gas borboteado, dando como resultado una solución relativamente libre de gases. Cuando se está usando el borboteo por helio hay que ser consciente de los potenciales cambios que pueden tener lugar en la composición de la fase móvil debido a la volatilidad selectiva de cualquier aditivo de la fase móvil, p.ej., ácido Trifluoroacético (TFA). Se debe evitar un borboteo por helio “vigoroso” ya que puede provocar perturbaciones excesivas en la superficie de la fase móvil, dando como resultado la introducción de más aire que el que es eliminado. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 37 - Incidencias y Mantenimiento en HPLC Desgasificación por Vacío. La desgasificación por vacío se puede llevar a cabo de tres maneras: • • • En discontinuo Incidental Desgasificación en línea La desgasificación en discontinuo tiene lugar en un matraz a vacío que contendrá la fase móvil a la que se someterá un proceso continuo de vacío, bien generado por aspiración con agua, o simplemente con una bomba de vacío. La desgasificación incidental se lleva a cabo haciendo pasar la fase móvil a través de un filtro de membrana. Puesto que el líquido se microdispersa mientras está expuesto a un ligero proceso de vacío, se produce la desgasificación. La desgasificación incidental es más bien un tratamiento secundario cuando se necesite eliminar materia particulada de la fase móvil por vacío antes de su uso. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 38 - Incidencias y Mantenimiento en HPLC La desgasificación en línea es el método más empleado actualmente. La fase móvil está encerrada en un tubo permeable a los gases mientras atraviesa la unidad de desgasificación. El gradiente de vacío generado hace expulsar los gases retenidos en la fase móvil, atravesando éstos la membrana del tubo. Ultrasonidos Este es el menos eficiente de todos los mecanismos de desgasificación ya que solo elimina aproximadamente el 30% de los gases disueltos. La fase móvil se desgasifica por sonicación de forma que los gases disueltos se aglomeran formando burbujas con el suficiente tamaño para flotar hasta la superficie del líquido. Dicha técnica es limitada, pues se lleva a cabo fuera del sistema y el calor generado en el proceso de sonicación puede provocar la pérdida de los compuestos más volátiles de la fase móvil por evaporación selectiva. Sus limitaciones se compensan generalmente por sus ventajas en simplicidad y bajo coste. En consecuencia el ultrasonidos no es muy recomendable y si es usado debe serlo durante el mínimo tiempo posible y preferiblemente con un baño de agua que controle la temperatura. La figura anterior muestra la eficacia relativa de los diferentes métodos de desgasificación, tomando como criterio el porcentaje de oxígeno eliminado de un volumen de metanol determinado en función del tiempo. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 39 - Incidencias y Mantenimiento en HPLC 3.4 Filtración de la Fase Móvil La filtración elimina el polvo además de otras partículas y contaminantes, siendo por tanto una etapa fundamental en la preparación de cualquier fase móvil tampón. Las partículas pueden alojarse en el sistema de aporte de disolvente, provocando desgaste en los componentes del cabezal de la bomba, generando fugas, flujos irregulares y cambios en la composición de la fase móvil. Las partículas pueden también alojarse en la válvula de inyección generando fugas cruzadas entre los puertos de la válvula que dan como resultado áreas y/o alturas de picos irreproducibles y causan una disminución de la vida útil de ésta. La filtración de la fase móvil también ayuda a prevenir la obstrucción de la línea, o de la guarda columna si la hubiere, evitando los cambios continuos de éstas y reduciendo pérdidas de tiempo y costes. El filtro en línea debe instalarse después del sistema de inyección pero antes de la columna analítica. De este modo cualquier partícula generada por el desgaste de los componentes del asiento del rotor procedentes de la válvula de conmutación del inyector será retenida de modo eficaz. Sin filtración de la fase móvil y sin añadir un sistema protector de filtro en línea, las partículas que entran en el HPLC pueden acumularse en cabeza de columna traduciéndose en una amplia variedad de problemas cromatográficos. Un bloqueo parcial en la entrada de la columna se traduce en un aumento de la presión y por tanto en deterioro de la forma de los picos cromatográficos, efecto que queda patente en desdoblamientos, frentes o colas en los picos. La figura siguiente muestra el efecto de una frita parcialmente bloqueada a la introducción de muestra en la columna. Sin bloqueo, la muestra sale de los tubos de conexión, distribuyéndose uniformemente dentro de la frita de entrada y después en cabeza de columna. Cuando hay un bloqueo parcial se impide la distribución homogénea de la muestra en cabeza de columna, obteniéndose anomalías en las formas de pico. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 40 - Incidencias y Mantenimiento en HPLC En ocasiones también puede haber cambios en los tiempos de retención del analito (TR), el factor de capacidad del analito (k’), disminución en la selectividad de la separación, presencia de picos falsos o adsorción irreversible del analito en la columna con la consecuente reducción de la vida útil de esta. Usar filtros mezcla de éster celulosa − acetato y nitrato de celulosa − (0.22µm de tamaño de poro) para disolventes acuosos tamponados, p.ej. Whatman Membra-Fil. Usar Teflon (Polytetrafluoroetileno − PTFE) (0.22µm de tamaño de poro) para solventes orgánicos. Es frecuente que la membrana de PTFE esté laminada sobre una trama de polipropileno como soporte para aumentar su durabilidad. Es muy recomendable reservar material de vidrio específico que solo se utilice para el proceso de filtración de la fase móvil y así prevenir contaminación imprevista. Un conjunto de vidrio debe dejarse solo para solventes acuosos tamponados y otro distinto para solventes orgánicos. Cualquier vidrio debe ser inspeccionado ante la posible presencia de material particulado para prevenir contaminación accidental de las fases móviles pre-filtradas Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 41 - Incidencias y Mantenimiento en HPLC Es una buena práctica el filtrar cualquier disolvente que no sea de grado para HPLC antes de mezclarlo y emplearlo en la preparación de fase móvil. Cuando se filtran mezclas de disolventes, las diferencias entre las presiones parciales de cada solvente pueden ser causa de cambios en la composición. En consecuencia, el usuario debe ser consciente de los cambios en la concentración de los solventes - y ser consecuente con los PNT (Procedimientos Normalizados de Trabajo). Generalmente no es aconsejable filtrar los disolventes de calidad HPLC suministrados por el fabricante. Estos disolventes se suministran ya eficazmente filtrados a 0.22µm y posteriormente embotellados bajo condiciones de “sala-limpia”. En consecuencia, el riesgo de introducción de materia particulada o contaminantes aumenta considerablemente cuando se realiza de nuevo la filtración en el laboratorio. Los filtros pueden estar revestidos o contener materiales como tensioactivos o glicerina, que podrían arrastrarse como contaminación a la fase móvil. Por tanto, es una buena práctica pre-lavar los filtros y desechar los primeros lavados. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 42 - Incidencias y Mantenimiento en HPLC Filtros de Disolventes en línea El uso de disolventes de alta calidad para HPLC suele eliminar la necesidad de filtración manual. Sin embargo, la mayoría de fabricantes recomiendan el uso de filtros para disolventes en línea: Generalmente los filtros están hechos de vidrio sinterizado o acero inoxidable, que deben ser limpiados o reemplazados mensualmente. El bloqueo de uno de los filtros causará pequeños cambios en la composición de la fase móvil, provocando fluctuaciones en los tiempos de retención: Fluctuaciones en los tiempos de retención debido al bloqueo de un filtro en línea. El mejor método de limpieza para los filtros de acero inoxidable es por ultrasonidos en isopropanol (IPA). Los filtros de vidrio sinterizado nunca deben ser lavados por ultrasonidos, siendo el mejor método la inmersión en IPA durante 24 horas. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 43 - Incidencias y Mantenimiento en HPLC Crecimiento microbiológico Los disolventes contaminados o el crecimiento microbiano en las botellas de disolvente, pueden taponar los filtros y perjudicar al funcionamiento de la bomba. El crecimiento microbiano también puede bloquear el automuestreador, provocando variaciones en los volúmenes inyectados o recubriendo las ventanas de la celda de flujo provocando así líneas de base erráticas y reduciendo la sensibilidad. Una línea de base errática causada por crecimiento microbiano en la fase móvil Es recomendable tomar las siguientes precauciones: • Si es posible usar botellas estériles para los disolventes. • Reemplazar disolventes acuosos y tamponados cada dos días. • Evitar la exposición directa al sol o usar botellas de vidrio ámbar. Otras Consideraciones para la Fase Móvil: Rango del pH Rango del pH del equipo 2.3 – 9.5 Rango del pH extendido 2.3 – 12.5 Corrosivos para el acero inoxidable Ácido clorhídrico Ácidos inorgánicos y ácidos fuertes Haluros Básicos (Cloruro de Sodio, Ioduro de Litio) Tetracloruro de Carbono con 2-propanol o THF Agentes acomplejantes (EDTA, ácido cítrico, ácido acético) Ataque al cuarzo y vespel Soluciones alcalinas, pH>11 Si se usan los compuestos anteriores será necesario un mantenimiento más frecuente del HPLC. Cuando se usen, al finalizar el análisis la bomba u otros componentes del HPLC deben ser purgados a fondo. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 44 - Incidencias y Mantenimiento en HPLC Otra cuestión relativa al uso de filtros es la filtración de las muestras antes de inyectarlas. Aunque filtrar la muestra permite eliminar partículas, existe la posibilidad de perder analitos por ser absorbidos irreversiblemente en la membrana de los filtros. El efecto inmediato sería la obtención de áreas y/o alturas de picos irreproducibles. Por tanto, es aconsejable realizar de una serie de pruebas previas que controlen la recuperación de los analitos a los niveles de concentración esperados cuantificando a partir de soluciones “fortificadas”, a fin de determinar si el filtro es compatible y no retiene los compuestos en cuestión. Dicho proceso se ilustra a continuación: Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 45 - Incidencias y Mantenimiento en HPLC 4 Sistemas de bombeo El objetivo de esta sección es: • • • • Describir la función de la bomba Describir los principios de funcionamiento del sistema de bombeo Describir los dos procesos por los cuales se forma el gradiente de fase móvil Enumerar y describirlas incidencias y los procesos de mantenimiento más comunes 4.1 Funciones de la Bomba La misión de la bomba es enviar continuamente un flujo fase móvil libre de pulsos al inyector, columna y detector, independientemente de la contrapresión del sistema. Cada sistema de bombeo debe cumplir los siguientes requisitos: • • • • • Todas las partes de la bomba en contacto con fluidos han de ser de materiales inertes El rango normal de presiones en HPLC es de 100 – 6000 psi (14.2 psi = 1 Bar) dependiendo del equipo utilizado. Esto correspondería a unos flujos de rango 0.5 - 10mL/min, dependiendo de la geometría y relleno de la columna, etc. La mayoría de las bombas están preparadas para alta presión, pero se utiliza un sistema limitador de presión (“Cut-off”) para evitar daños en otros componentes del sistema como la celda de flujo del detector. Algunas bombas de tipo flujo constante pueden producir un perfil de presión pulsado. Si el detector usado es sensible al flujo se puede producir ruido u ondulaciones de la línea de base. Tales variaciones de flujo son normalmente “amortiguadas” usando un volumen muerto comprimible situado entre la bomba y el inyector (“Damper”). El fabricante normalmente incorpora el “dámper” en el propio módulo de bombeo. El volumen interno de la bomba debería ser lo más pequeño posible. Esto garantiza que los cambios en la composición de la fase móvil puedan ser rápidos durante el gradiente de elución. Se evita de esta forma que los tiempos de re-equilibrio resulten el paso limitante en cualquier análisis. Los sellos de las bombas, arandelas, juntas y ocasionalmente los pistones del cabezal de la bomba necesitan ser reemplazados, por lo que deben ser de fácil acceso. Los asientos de las válvulas antirretorno tienen tendencia a desgastarse y necesitarán sustituirse si se bloquean o quedan pegados. La forma más efectiva de prolongar el tiempo entre mantenimientos es filtrar la fase móvil y usar unos procedimientos correctos de puesta en marcha y apagado Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 46 - Incidencias y Mantenimiento en HPLC 4.2 Bomba Recíproca de Pistón Simple El mayor reto de cualquier fabricante de bombas de HPLC es proporcionar un flujo de fase móvil libre de pulsaciones. La simplicidad del cabezal de la bomba de pistón simple es ideal para separaciones isocráticas, que requieren un flujo fijo de un único solvente durante todo el análisis. La incorporación de un amortiguador de pulsos (“damper”) de bajo volumen permite obtener un flujo libre de pulsaciones. Además de minimizar las pulsaciones de flujo gracias a la incorporación de un amortiguador de pulsos, los equipos de HPLC modernos pueden variar la velocidad del pistón del cabezal de la bomba, de forma que su velocidad sea menor en la etapa de dispensación que en la etapa de llenado del pistón, minimizando de esta forma los efectos de la pulsación. Principio de Operación Las válvulas antirretorno (“Check-Valves”) ó válvulas de “Bola y Asiento” están diseñadas para permitir únicamente un flujo unidireccional de fase móvil. La “bola”, hecha de un material químicamente inerte como el rubí, se asienta en un asiento de zafiro sintético. En función de la posición del pistón en un determinado momento la bola se encuentra situada directamente contra el asiento, deteniendo de forma efectiva el flujo de la fase estacionaria, o suspendida en el cuerpo principal de la válvula, donde ofrece poca resistencia al flujo de la fase móvil, permitiendo su paso al sistema de HPLC. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 47 - Incidencias y Mantenimiento en HPLC Las imágenes ilustran el funcionamiento de las válvulas de entrada y salida durante los periodos de llenado y dispensación de una bomba recíproca de pistón simple. Aunque las válvulas antirretorno proporcionan miles de horas de funcionamiento ininterrumpido, son componentes consumibles, y como tales requerirán de limpieza o sustitución después de un uso prolongado. Las fases móviles sin filtrar, corrosivas o altamente tamponadas reducirán considerablemente el tiempo de vida de las válvulas antirretorno. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 48 - Incidencias y Mantenimiento en HPLC 4.3 La Bomba Recíproca de Doble Pistón Comparándola con la bomba de pistón simple, el diseño de la bomba recíproca de doble pistón ofrece flujos más reproducibles con la correspondiente reducción de las fluctuaciones de presión. Sin embargo, la mayor complejidad del diseño de la bomba de doble pistón aumenta también sus requerimientos de mantenimiento, puesto que el pistón adicional y los sellos del mismo están sometidos también a desgaste y requieren sustitución. Principio de Operación Este diseño de la bomba es muy eficiente, porque mientras un pistón se está llenando con fase móvil, el segundo pistón está entregando fase móvil al sistema de HPLC. Este desfase de 180º en el funcionamiento del pistón produce un flujo de fase móvil virtualmente libre de picos, y permite gran reproducibilidad en gradientes de fase móvil. Este diseño se utiliza a menudo junto con un amortiguador de pulsos, para obtener la menor fluctuación de presión posible durante la dispensación de la fase móvil. La imagen ilustra el principio de funcionamiento de la bomba recíproca de doble pistón. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 49 - Incidencias y Mantenimiento en HPLC 4.4 La Bomba Binaria de Gradiente Las bombas isocráticas sirven para atender las necesidades básicas del cromatografista moderno. Sin embargo, cuando se requiere un gradiente de fase móvil, durante el cual se introduce gradualmente en la fase móvil un solvente más fuerte para eluir de la columna los analitos fuertemente retenidos, se requiere un diseño de bomba más sofisticado. Hay dos formas básicas de conseguir un gradiente en la fase móvil. La primera es emplear dos bombas reciprocas trabajando al unísono –esto se denomina bomba binaria de gradiente. Las bombas binarias consisten en dos “canales” de solvente, donde cada canal está conectado a una bomba recíproca idéntica y el flujo de disolvente procedente de cada bomba se combina en una cámara de mezcla de bajo volumen, un amortiguador de pulsos, y finalmente una segunda cámara de mezcla. Los flujos individuales de bombeo –o ratio volumen/flujo- de cada una de estas bombas recíprocas están controlados electrónicamente, permitiendo mezclar proporcionalmente el solvente de los dos canales. Por lo tanto, si se necesita un gradiente con la composición de 50% del solvente A y 50% del solvente B, las bombas trabajarán a flujos iguales, y si el flujo total deseado es de 1mL/min, cada bomba entregará el solvente a 0.5mL/min. Puesto que la mezcla de solventes se produce después de las bombas, se denomina “mezcla a alta presión”. La imagen ilustra el principio de operación de la bomba de gradientes de doble pistón recíproco Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 50 - Incidencias y Mantenimiento en HPLC 4.5 La Bomba Cuaternaria de Gradiente Una solución alternativa para la formación de un gradiente de fase móvil es usar solamente una bomba de doble pistón conectada a una válvula proporcional de cuatro canales controlada por el software. La válvula proporcional se encuentra entre el desgasificador y la bomba, y permite introducir volúmenes exactos de cada solvente en una cámara de mezcla, suministrando después la mezcla de solventes formada al sistema de HPLC. Cada puerto de la válvula solenoide permanece abierto sólo una fracción de lo que se denomina “ciclo de trabajo” de la bomba. El gradiente de elución se consigue alternando la fracción de este ciclo en que cada solenoide permanece abierto, permitiendo que la fuerza de elución efectiva de la fase móvil cambie en función del tiempo. Por lo tanto, si el ciclo de la bomba es de un segundo y la composición del gradiente deseada es del 25% de solvente A, 25% del solvente B, 25% del solvente C y 25% del solvente D, cada solenoide permanecerá abierto un cuarto de segundo, permitiendo a las alícuotas de cada uno de los cuatro solventes mezclarse en igual proporción. Las características individuales de viscosidad y compresibilidad de cada solvente afectarán al funcionamiento de la válvula de proporción, y pueden ajustarse convenientemente desde el software del instrumento. En comparación con la bomba de gradiente binario, donde la mezcla del solvente ocurre después de la bomba y se denomina “mezcla a alta presión”, la bomba de gradiente cuaternario mezcla el solvente antes de la bomba y se denomina “mezcla a baja presión”. Esta mezcla no es tan efectiva como la producida a alta presión, y muestra: • Menor reproducibilidad del gradiente Cuando se trabaja a flujos menores de 1.0mL/min p.ej., las bombas de gradiente binario ofrecen mayor precisión y reproducibilidad que las bombas cuaternarias. Además, si existe una diferencia apreciable en la proporción relativa de los solventes mezclados, por ejemplo del 98% de solvente A y 2% del solvente B, de nuevo se obtendrá mayor reproducibilidad con un sistema de mezcla a alta presión que con uno que emplee válvula de mezcla a baja presión. • Efectos de Cavitación Durante la mezcla de disolventes en la válvula proporcional pueden generarse burbujas. Esto es debido a que los gases disueltos tienen menor solubilidad en la mezcla final que en los solventes iniciales. • Líneas de base ruidosas y erráticas Durante la mezcla a baja presión podría producirse inmiscibilidad entre los disolventes y formación de burbujas. Estos efectos producen caídas de presión en la mezcla, variación de los flujos, y contribuyen a líneas de base erráticas y ruidosas. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 51 - Incidencias y Mantenimiento en HPLC La imagen muestra el funcionamiento de la válvula proporcional en una bomba recíproca de doble pistón 4.6 Formación de Gradientes y Tiempo Muerto (“Dwell-Time”) El volumen interno de una bomba de HPLC debería ser idealmente lo más pequeño posible, esto permite una rápida formación del gradiente y además una transferencia efectiva de los métodos con gradiente entre sistemas intra- e inter-laboratorio. El tiempo que se necesita entre la formación del gradiente de solvente hasta que éste llega al comienzo de la columna analítica se denomina “tiempo muerto del sistema” (tD). El tiempo muerto del sistema se calcula determinando primero el “volumen muerto del sistema (VD), que es característico del volumen extra-columna mostrado por la bomba y la longitud total de los conectores entre el HPLC, y se determina como sigue: Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 52 - Incidencias y Mantenimiento en HPLC Cálculo del Volumen Muerto del Gradiente 1. Quitar la columna y conectar el inyector al detector mediante una pequeña unión de 0.010-in. d.i. (o menor) – esto se suele denominar “capilar de restricción”. Un regulador de contrapresión después del detector ayudará a mantener la suficiente presión en el sistema para un buen funcionamiento de la válvula antirretorno (“Check-Valve”). 2. Como solvente A usar agua calidad HPLC, y como solvente B añadir alrededor de 0.1% de acetona a agua HPLC (también puede usarse metanol o acetonitrilo en lugar de agua). 3. Ajustar la longitud de onda a la máxima absorbancia del compuesto añadido (265nm para la acetona). 4. Realizar un cromatograma a 100% de A y otro a 100% de B, ajustando la escala de absorbancia en los datos del sistema, para que ambas señales del solvente muestren una señal dentro de la escala 5. Programar un gradiente lineal de 0-100% B en 10min a 2mL/min. Las condiciones exactas no son críticas, simplemente asegúrese que el volumen total del gradiente es al menos de 20mL. Mantenga una etapa isocrática al 100% B durante al menos 5min al final. El cromatograma resultante debería aparecer aproximadamente como en la figura superior. La etapa isocrática al principio de la gráfica representa el volumen muerto del sistema (VD), seguido por la etapa de gradiente, y finalmente la etapa isocrática mantenida hasta el final. La curvatura del cromatograma debería ser mínima al final del gradiente, y debería ser lineal excepto al principio y al final. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 53 - Incidencias y Mantenimiento en HPLC 6. Determinar en primer lugar el tiempo de retención del sistema situando el tiempo a mitad de gradiente (t1/2) – la mitad de la distancia vertical entre el inicio y el final de los segmentos isocráticos; 9.2min para el cromatograma descrito. Restar a continuación la mitad del tiempo de gradiente; 10min/2 = 5min para este ejemplo de (t1/2) El resultado es el tiempo muerto del sistema (tD); 9.2min – 5min = 4.2min. Finalmente transformar tD en volúmen muerto (VD) multiplicando por el flujo empleado en el método p.ej. 4.2min x 2mL/min = 8.4mL. Es esencial que los tiempos de equilibrio de la columna y del sistema se controlen durante el re-equilibrio del gradiente. Esto es, en este ejemplo, el tiempo total de equilibrio usando una columna de 150 x 4.6mm a un flujo de 2mL/min sería: 4.2 min (tD) + (1.7ml (volumen muerto de la columna) / 2ml/min x número de volúmenes de columna) O para un re-equilibrio de 5 volúmenes de columna: 4.2 + ((1.7/2) x 5 = 8.45min O para un re-equilibrio de 10 volúmenes de columna: 4.2 + ((1.7/2) x 10) = 12.7min Los sistemas de mezclado a alta presión con bombas binarias pueden conseguir volúmenes extra-columna muy pequeños. Las mezclas a alta presión proporcionan los menores volúmenes muertos ya que el gradiente se forma después de la bomba, esto se traduce en un menor tiempo de retención cuando se requieren gradientes rápidos o muy rápidos. Quitar la segunda cámara de mezcla reduce aún más el volumen de retención. El volumen efectivo de mezcla de un sistema de gradiente cuaternario a baja presión necesita ser mayor que para un sistema de gradiente binario a alta presión para asegurar una correcta mezcla de solventes. Este hecho incrementa el volumen muerto, haciendo que la respuesta de estos sistemas se alargue y los cambios en el gradiente se muestren apreciablemente más lentos. Ajustar el volumen de retención (VD) a su HPLC le permitirá una transferencia del método más eficaz. Compensando los diferentes volúmenes muertos en diferentes sistemas, los tiempos de retención de los analitos y por lo tanto el perfil del cromatograma pueden mantenerse idénticos. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 54 - Incidencias y Mantenimiento en HPLC 4.7 Incidencias de la Bomba La Válvula de Purga La causa más probable de un incremento de presión en el sistema es un bloqueo parcial en la primera frita después del sistema de inyección – El filtro In-Line o la pre-columna- sin embargo si ninguno de estos dos elementos están instalados, el problema puede venir de la cabeza de la propia columna. Si tales aumentos de presión son comunes, es recomendable seguir algunas medidas preventivas adicionales, como instalar un filtro de línea o someter a las muestra a una filtración previa antes de su inyección. El conjunto de válvula de purga contiene PTFE como frita en la mayoría de los sistemas de HPLC. Está diseñado para eliminar los depósitos que puedan provenir de los sellos de los pistones arrastrados por la fase móvil. Cuando esta frita se bloquea se produce un incremento en la contra presión del sistema. Si esta presión llega aproximadamente a los 10 Bar mientras se bombea fase móvil con la válvula de purga abierta, debe cambiarse la frita No sustituir la frita puede dar como resultado una línea de base errática Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 55 - Incidencias y Mantenimiento en HPLC El material del sello del Pistón puede ser arrastrado hasta bloquear la frita y la parte superior de la columna analítica. Los bloqueos en este punto retrasan la entrada de los analitos en la columna, produciendo picos no uniformes con hombros. Este tipo de picos también pueden originarse por otro tipo de anomalías, pero una frita de columna bloqueada, a menudo provocará hombros en todos los picos cromatográficos Una frita de Columna bloqueada por restos del sello de los pistones produciendo picos con hombro Pistón y Sellos Los sellos del Pistón están diseñados para desgastarse con el uso, siendo su vida útil muy dependiente del tipo y de la concentración de las fases móviles tampón que se utilicen. Una fuga en el sello del pistón provoca fluctuaciones en la línea de base como podemos observar en el gráfico: Línea de base sinusoidal indicativa de un pistón rallado o sello con fugas Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 56 - Incidencias y Mantenimiento en HPLC Cualquier cambio del sello del pistón deberá implicar una inspección visual del mismo, ya que si existen arañazos en la superficie del pistón, pueden llevar prematuramente al fallo del nuevo sello reemplazado. Antes de la inspección del pistón se deben limpiar los posibles depósitos de sales, principalmente los restos de los tampones o del sello, utilizando un trapo limpio, sin pelusas, empapado con una mezcla 1:1 de H2O:MeOH , y solo en una dirección , desde la base hacia el extremo. Luego, lo examinaremos con luz brillante y con la ayuda de una lupa para verificar que no hay presente ningún arañazo o estrías. Los sellos del Pistón están diseñados con frecuencia para deformarse con el fin de ajustarse al pistón con el que están emparejados. En consecuencia es posible que haya que sustituir el pistón y el sello al mismo tiempo. Nota: Algunos fabricantes de instrumentos ofrecen e incluyen accesorios integrados para la limpieza continua de los sellos facilitando así la eliminación de los depósitos de cristal que proviene de los tampones alrededor de los sellos. Con estos accesorios se hace pasar una solución de lavado por los sellos de pistones para aumentar la vida útil de los mismos. Una buena solución para lavado es la formada por un 10% de isopropanol en agua. En algunos casos esta solución de lavado también sirve para lubricar el movimiento del pistón a través de un segundo conjunto de sellos residente en el propio hardware del sello. En ese caso debe usarse siempre una solución de lavado, incluso aunque no se empleen tampones como fase móvil. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 57 - Incidencias y Mantenimiento en HPLC Válvulas Antirretorno La cristalización de los tampones, el crecimiento microbiano en las botellas de los disolventes y el aire pueden perjudicar el sellado entre la bola de rubí y el soporte o asiento de zafiro en la válvula antirretorno. La sedimentación y el crecimiento microbiológico pueden provocar que este problema permanezca constantemente. En estos casos puede observarse una línea base con fluctuaciones características y repetitivas: Típica línea de base producida por una válvula antirretorno defectuosa Para regenerar una válvula antirretorno pegada o parcialmente bloqueada, se puede seguir el siguiente proceso de limpieza: Sonicar durante 10mins en una mezcla 1:1 de H2O:MeOH (1:1) Sonicar durante 10mins en 0.1M HNO3. Sonicar durante 5mins in MeOH antes de reinstalarla Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 58 - Incidencias y Mantenimiento en HPLC Burbuja(s) de Aire en el Cabezal de la Bomba La captura de aire es un problema relativamente común en bombas de pistón recíproco, produciéndose en aquellos componentes del sistema que están contacto directo con la fase móvil como las válvulas de entrada y salida, pistón, sello del pistón, y cabezal de la bomba. Las burbujas en el interior de los cabezales de la bomba a menudo generan una línea de base de tipo sinusoidal: Las burbujas de aire, si no se introducen directamente en el sistema por una deficiente desgasificación de la fase móvil, pueden formarse por la creación de un ligero vacío en el cabezal durante el ciclo de succión (llenado) de la bomba. La combinación de baja presión, permitiendo la desgasificación, y los “puntos de nucleación” de la superficie de de los componentes son ideales para la creación de burbujas. La válvula proporcional, utilizada para la mezcla en los sistemas a baja presión para la creación las condiciones isocráticas o en gradiente requeridas, puede también introducir burbujas debido a la menor solubilidad del aire en la mezcla de solventes comparada con la de los solventes puros individuales. Este efecto se denomina “cavitación”. Una vez que la pequeña burbuja se ha formado puede actuar en realidad como un amortiguador de presión, amortiguando el efecto de succión y dispensación de la bomba, haciendo que ésta proporcione un flujo bajo y a menudo errático. Aflojar ligeramente las conexiones de las válvulas de salida y dejar fluir fase móvil del cabezal de la bomba puede servirnos para purgar las burbujas de aire. En la mayoría de las ocasiones simplemente abrir la válvula de purga y aumentar el flujo de fase móvil - a flujos de hasta 5mL/min- será suficiente para eliminar las burbujas de aire presentes. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 59 - Incidencias y Mantenimiento en HPLC Una disminución de la presión de trabajo generalmente implica una fuga en el sistema. Debe corregirse apretando, reajustando o cambiando el conector defectuoso. Los fabricantes a menudo incorporan un test específico para el diagnóstico de fugas, que puede realizarse para verificar la integridad del cabezal de la bomba y otros componentes del sistema. El fallo en alguna parte del test puede relacionarse con el mal funcionamiento de algún componente específico del sistema cromatográfico reduciéndose así el tiempo en que el instrumento queda fuera de operación al detectarse más rápidamente la avería. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 60 - Incidencias y Mantenimiento en HPLC Tutorial 1 Pregunta 1 Del siguiente listado de componentes selecciona aquellos que contribuyen a un ensanchamiento de las bandas cromatográficas, es decir, disminuyen la eficiencia en un sistema típico de HPLC Componente HPLC Si/No Tubos de conexión Filtro de Línea Cabezales de la bomba Guarda-columna Inyector Conexiones-uniones Celda de flujo del Detector Botellas de disolvente Columna Analítica Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 61 - Incidencias y Mantenimiento en HPLC Pregunta 2 La ilustración siguiente es el diagrama esquemático de un cabezal de doble pistón recíproco. Identifica y etiqueta cada componente. Usando la tabla siguiente, explica cual es la función de cada uno de los componentes e indica cuál es su fallo asociado más frecuente Componente del Equipo Función Incidencia Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 62 - Incidencias y Mantenimiento en HPLC Pregunta 3 Evaluar el problema propuesto en el siguiente cromatograma y sugerir las posibles causas y cómo podemos corregirlo Pregunta 1 Observaciones: Posible(s) Causa(s) y Solución(es): Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 63 - Incidencias y Mantenimiento en HPLC Pregunta 4 Un gas disuelto en la fase móvil puede provocar múltiples problemas cromatográficos. Considera éstos y completa la siguiente tabla. Problema Observado Causa Acción Correctora Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 64 - Incidencias y Mantenimiento en HPLC 5 Inyectores El Objetivo de esta sección es: • • • Describir la función del sistema de inyección del HPLC. Explicar los principios de operación de los sistemas de inyección HPLC más comunes. Enumerar y describir las incidencias más frecuentes y los procedimientos de mantenimiento. 5.1 Componentes del Sistema de Inyección El sistema de inyección se encuentra situado después de la bomba, estando por lo tanto localizado en la parte alta presión del sistema de suministro de solventes. La inyección de una muestra (a baja presión) en el sistema representa una etapa crítica del proceso cromatográfico. Las válvulas de inyección de muestra, o válvulas de conmutación, permiten la introducción de cantidades precisas de muestra en el sistema y en la columna. Inyectando directamente en el flujo de fase móvil a alta presión se elimina la necesidad de parar el flujo de fase móvil, minimizando así de forma efectiva cualquier perturbación en la línea de base cromatográfica realizando un cambio, esencialmente libre de pulsaciones, en la dirección del flujo de fase móvil. La figura siguiente muestra la válvula de conmutación Rheodyne de 6 puertos y dos posiciones, cuyo diseño conforma la base para todas las válvulas de inyección. La válvula se compone de una parte estática (“stator”) enfrentada a un sello giratorio (“rotor seal”), en el que se han mecanizado unos canales entre los puertos que permiten que el flujo de fase móvil evite el loop(*) de inyección (“by-pass”) para la carga de muestra (“load”), o vaya hacia él (“inject”) para la inyección de la muestra. En la posición intermedia entre “load” e “inject”, el sello del rotor bloquea momentáneamente el flujo de fase móvil hacia la columna. Este bloqueo causa un pulso de presión en cabeza de columna, porque por un tiempo finito el flujo efectivo de fase móvil cae a cero. Puesto que tales pulsos no son deseables, la válvula debe girarse lo más rápidamente posible para minimizar cualquier pulso de flujo y presión. Además, estos repetidos choques de presión pueden dañar la columna, comprimiendo su fase estacionaria y provocando la aparición de volúmenes muertos que producirán formas de pico cromatográfico defectuosas, ejemplificadas por la aparición de hombros y desdoblamiento en los picos. (*) La traducción de LOOP es BUCLE pero ha preferido mantenerse la palabra inglesa original por la generalización de su uso en el argot cromatográfico. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 65 - Incidencias y Mantenimiento en HPLC Componentes de una válvula de conmutación Rheodyne de 6 puertos El Rotor es un disco fabricado normalmente en material polimérico Vespel. Esta poliimida confiere al rotor unas características de bajo desgaste y alta Resistencia, admitiendo un rango de pH de 0−10. Figura mostrando rotores de inyección de Vespel . Para fases móviles con pH sobre 10 se recomienda un Rotor Seal compuesto de material polimérico Tefzel o PEEK, ya que ambos proporcionan mayor resistencia química y versatilidad en el rango completo de pH de 1−14. Los ácidos oxidantes fuertes, como el Ácido Nítrico o el Sulfúrico no son compatibles con PEEK. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 66 - Incidencias y Mantenimiento en HPLC Figura mostrando el rango de pH de los diferentes materiales NB: los colores de pH mostrados son sólo con fines ilustrativos El rotor está situado internamente en el cuerpo de la válvula de inyección, y hace un cierre a alta presión contra la cara del estátor. Como el rotor gira físicamente en cada secuencia de inyección, y sufre un deterioro de los canales entre los puertos por la exposición a un pH alto y por las raspaduras producidas por la fase móvil o partículas contenidas en los solventes, se desgastará con el uso. En consecuencia, este es uno de los componentes del HPLC que necesitarán una sustitución rutinaria. La cara del estátor y el cabezal permiten la introducción y expulsión de la fase móvil a través de los puertos capilares específicos de entrada y salida. El puerto de inyección facilita el llenado del loop de muestra con una jeringa, mientras que el puerto de desechos (“waste”) ventea el loop permitiendo que se elimine el exceso de muestra. Existen varias configuraciones para la inyección de un determinado volumen de la solución de muestra en la fase móvil; uno, muy ampliamente utilizado emplea un loop de inyección de volumen pre-determinado. Este loop puede llenarse de dos formas distintas: Llenado Completo del Loop Puesto que el loop contiene fase móvil, la solución de muestra introducida se mezclará y se diluirá necesariamente con esta fase móvil residente mientras la desplaza. Por tanto, para que el loop se rellene de forma homogénea, debe inyectarse un volumen de solución de muestra de entre 2 y 5 veces el volumen del loop, eliminando así cualquier posible efecto de dilución. Aunque el loop controle efectivamente el volumen de muestra inyectada, haciendo que la cantidad de muestra introducida por la jeringa no sea un parámetro critico, para obtener máxima precisión y linealidad es una práctica recomendada cargar siempre el loop con el mismo volumen de muestra p.ej. 3x volumen del loop ± 10%, En consecuencia, para un loop de 100µL se correspondería con un volumen de 270−330µL por inyección. La Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 67 - Incidencias y Mantenimiento en HPLC reproducibilidad es esencial para mantener la precisión y exactitud; El volumen absoluto inyectado es de menor importancia. Llenado Parcial del Loop La técnica de llenado parcial del loop se utiliza habitualmente cuando es importante ahorrar una muestra valiosa o limitada. Para obtener la mejor precisión posible el volumen de muestra inyectada no debe superar el 50% de la capacidad del loop del inyector. Durante la carga del loop, la muestra introducida “empuja” a la fase móvil de su interior enviándola fuera a través de la salida de desechos (“waste”), y durante ese proceso de intercambio el frente de muestra se diluye. Este efecto de dilución está causado por el perfil de flujo parabólico de un líquido en movimiento por el interior de un segmento de tubo ya que debido al flujo laminar, el centro de la corriente de fluido se mueve más deprisa que la capa de líquido que está cerca de la pared del tubo. Una forma de evitar la citada discriminación durante la inyección es inyectar una burbuja de aire inmediatamente delante del volumen de muestra inyectado. De esta forma se evita cualquier posible mezcla de la solución de muestra con la fase móvil contenida en el loop. La consecuencia de este fenómeno es que la muestra diluida ocupa en realidad unos 2µL del volumen del loop por cada 1µL de la muestra cargada con la jeringa. Por lo tanto, asegurándonos de cargar <50% del volumen efectivo del loop, evitamos que el frente de la banda de muestra diluida salga del loop y provoque errores relacionados con la precisión y a la falta de reproducibilidad. Este efecto se muestra en el siguiente conjunto de figuras, que ilustra los efectos de la dilución de muestra en el área de pico obtenida: Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 68 - Incidencias y Mantenimiento en HPLC 1. Injección de 1µL de muestra en un loop de volumen fijo de 20µL. Los efectos de flujo laminar crean un tamaño de muestra diluida de 2µL. 2. Injección de 5µL de muestra en un loop de volumen fijo de 20µL. Los efectos de flujo laminar crean un tamaño de muestra diluida de 10µL. 3. Injección de 10µL de muestra en un loop de volumen fijo de 20µL. Los efectos de flujo laminar crean un tamaño de muestra diluida de 20µL. 4. Inyección de 20µL de muestra en un loop de volumen fijo de 20µL. El volumen de inyección es el mismo que un llenado completo del loop. Los efectos de flujo laminar crean un volumen de muestra ligeramente diluido. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 69 - Incidencias y Mantenimiento en HPLC La figura anterior muestra los efectos de dilución representando el área obtenida frente a los diferentes volúmenes de inyección en un loop de volumen fijo de 20µ µL. Cuando se carga un volumen <10µL de muestra existe una relación lineal entre el volumen introducido en el loop y la cantidad de muestra inyectada en columna. Cuando se cargan >40µL, el volumen real inyectado es ± 5% del volumen del loop. Sin embargo, en el rango entre 10−40µL se observa una relación no-lineal diferente. Después de la inyección son necesarios 5−10 volúmenes de loop para desplazar completamente la muestra del mismo. Se recomienda por tanto, mantener el inyector en la posición "inject" (paso principal) el tiempo necesario para permitir que la fase móvil fluya a través del loop arrastrando completamente la muestra. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 70 - Incidencias y Mantenimiento en HPLC 5.2 Automuestreadores Los automuestreadores son ampliamente utilizados en los laboratorios de análisis para aumentar el nº de muestras analizadas, mejorar la precisión, eliminar la necesidad de que haya personal presente para hacer las inyecciones manuales y reducir los costes laborales asociados al análisis rutinario. Aunque los fabricantes han diseñado automuestreadores bajo diferentes principios y estrategias, todos ellos incluyen cuatro componentes esenciales que permiten el automatismo mecánico del sistema de inyección manual: 1. 2. 3. 4. Las muestras se mantienen en viales de tamaño estándar. Cada vial está sellado con un septum, que, o bien es parte integral del tapón, o se ajusta en su lugar gracias a éste, evitando la evaporación selectiva del disolvente de la muestra y los consiguientes de cambios en su concentración. Los viales están situados en gradillas que permiten un orden de inyección aleatorio o en serie. Dichas gradillas pueden ser termostatizadas para evitar la degradación de las muestras térmicamente lábiles. Se emplea una aguja de inyección para penetrar el septum, y tomar el volumen de muestra requerido para la inyección. Dependiendo del automuestreador, dicha aguja puede ser fija o intercambiable. Una válvula de inyección commutable que permite la introducción de la muestra en el loop previamente a la inyección. La conmutación de dicha válvula de inyección se realiza bien por un actuador neumático o eléctrico. La mayoría de automuestreadores emplean alguna de las tres configuraciones de carga siguientes, en las que la muestra es o bien “extraída” o “empujada” hacia un loop de volumen fijo, o bien la aguja de inyección es parte integral del loop de inyección. 5.2.1 Automuestreador Llenado” de “Extracción- Este diseño de automuestreador extrae la muestra en la dirección del loop por succión con la jeringa. Debido a su simplicidad y fiabilidad fue muy popular, pero actualmente es redundante debido a los otros dos diseños. A continuación se muestra el esquema de operación de este tipo de automuestreador. Se une una jeringa al puerto de desechos y una aguja, por medio de tubos de conexión, al puerto de inyección. La aguja se inserta en el vial y extrae muestra de éste hasta que se ha llenado el loop con el volumen de inyección especificado. El giro posterior del rotor de la válvula de inyección permite arrastrar la muestra contenida en el loop hacia la cabeza de columna. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 71 - Incidencias y Mantenimiento en HPLC Load Position Inject Position Aunque es a la vez simple y fiable, este diseño de automuestreador adolece de una excesiva pérdida de muestra durante la secuencia de inyección. Como muestra la figura, es necesario mucho volumen de muestra para llenar la aguja y sus tubos de conexión antes de llegar al loop, y en consecuencia se pierden entre 10−100µL de muestra por inyección. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 72 - Incidencias y Mantenimiento en HPLC 5.2.2 Automuestreador de “Empuje-Llenado” En este diseño de automuestreador la aguja se mueve hacia el vial, toma el volumen requerido de muestra y se mueve entonces hacia el puerto de inyección para “empujar” la muestra al interior del loop. El llenado y vaciado de la jeringa se controla con un motor de pasos que es capaz de ofrecer una dispensación de volumen precisa y reproducible. Aunque generalmente queda un pequeño volumen residual de muestra en la aguja y su capilar de conexión, éste diseño no presenta el gran desperdicio de muestra mostrado por el diseño “extracción-llenado”, y en consecuencia requiere un menor volumen “extra” de muestra. Esta característica es una ventaja para el análisis de trazas, donde a menudo el volumen de muestra disponible es limitado. Load Position Wash Step Inject Position Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 73 - Incidencias y Mantenimiento en HPLC 5.2.3 Automuestreador de Loop Integrado Aunque es más complicado de operación que los dos diseños previos, las características del diseño del automuestreador de loop integrado aseguran que no haya pérdidas de muestra y como consecuencia de ello haya ido aumentando su popularidad. La serie de figuras mostrada a continuación ilustra la secuencia de eventos de carga e inyección de muestra: 1. Se dispensa y/o succiona un disolvente de lavado a través del loop, con la aguja asentada firmemente en un asiento de aguja de alta presión. 2. Después de lavar la aguja, ésta se mueve hacia el vial, donde se extrae el volumen especificado de muestra directamente hacia el loop por medio de un sistema de medida calibrado. El sistema de medida se compone de un pistón y su sello, que pueden desgastarse produciendo fugas y volúmenes de inyección no reproducibles. 3. La aguja se inserta de nuevo en el asiento de inyección de alta presión y el rotor de la válvula de inyección se mueve hacia su posición de inyección con lo que la muestra es arrastrada hacia la cabeza de columna. Puesto que la muestra se encuentra completamente contenida en la porción del loop barrida por la fase móvil, se inyecta toda la muestra que fue extraída del vial. Además, el lavado continuo de la válvula de inyección y el loop, que se produce tras la inyección, elimina los efectos de memoria (“sample carry-over”). El volumen máximo que puede inyectarse es función del tamaño del loop integrado, típicamente 100µL. Los fabricantes ofrecen también una función de multi-extracción (multidraw option) para compensar esta limitación en el volumen de inyección. Cuando se emplea esta opción se permite que varios volúmenes de 100µL sean combinados hasta un máximo de 1.400mL aumentando así el volumen de inyección y ayudando al análisis de trazas. La parte más débil de este diseño de automuestreador es el sello de alta presión, que tras las repetidas inserciones de la aguja eventualmente se irá desgastando y fugará siendo necesaria su sustitución. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 74 - Incidencias y Mantenimiento en HPLC La ruta que sigue la fase móvil en su paso por el automuestreador se denomina a veces “‘main pass’ o camino principal El sello del Rotor gira para permitir a la aguja que entre en el vial de muestra y al pistón que extraiga ésta hacia el loop. La Fase Móvil evita (“by-pass”) los principales componentes del sistema de inyección. Esta posición se conoce frecuentemente como posición de “bypass” o de carga”Load” Posición de Carga El Rotor vuelve a su posición inicial (“main pass”) y la Fase Móvil empuja la muestra contenida en el loop hacia la columna Posición de Inyección Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 75 - Incidencias y Mantenimiento en HPLC 5.3 Incidencias del Automuestreador La tabla siguiente muestra los problemas citados con más frecuencia sobre los automuestreadores: Problema % del Total Obstrucción de la aguja de muestra 22 Mala precisión y reproducibilidad 17 Problemas mecánicos 14 Posición de la Gradilla e Identificación de la muestra 12 Efecto Memoria 9 Fugas 9 Sellos 7 Prestaciones 6 Otros 5 Fuente − LC-GC Europe Volume 4, No.5 (1986) Pg. 416−420 La aguja de muestra La obstrucción de la aguja de muestra es el problema más frecuente que presentan los automuestreadores. Una preparación adecuada de la muestra ayudará por lo general a reducir el número de problemas creados por partículas, bien filtrando o centrifugando las muestras antes de su inyección. Para desbloquear una aguja obstruida se recomienda la siguiente secuencia de limpieza: 1. 2. 3. Empape la aguja con un disolvente apropiado p.ej. isopropanol. No sonique la aguja si está soldada al capilar ya que existe la posibilidad de que se fracture o se rompa por la unión. Intente desplazar la obstrucción con un alambre. Conecte la aguja a la bomba por medio de un capilar y haga fluir disolvente con presiones progresivamente crecientes para “reventar” la obstrucción. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 76 - Incidencias y Mantenimiento en HPLC El asiento y el capilar de alta presión Las fugas en esta zona son por lo general detectables a simple vista y bastante evidentes. El asiento de la aguja se deformará para ajustarse a la forma exacta de la aguja, por lo que la aguja y el asiento deberán cambiarse al mismo tiempo La muestra y la fase móvil comienzan a mezclarse justo después del asiento de aguja de alta presión. Este capilar es propenso a obstruirse bien por partículas de desolvatación de las muestras o de los tampones sólidos. El ruido esporádico (Spikes) en la inyección es un indicador de tales obstrucciones. El Rotor Seal El Efecto memoria, junto a una mala precisión en areas y alturas, son indicios de un rotor seal agotado. El rotor seal debe revisarse para buscar síntomas de desgaste, especialmente un ahondamiento en los microcanales entre los puertos que puede generar puntos de retención de muestra localizados. Estos puntos se “limpian” en la siguiente inyección produciendo un mini-cromatograma o picos fantasma. Un pico fantasma causado por contaminación en el rotor seal Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 77 - Incidencias y Mantenimiento en HPLC Figura mostrando la adsorción y efecto memoria de un rotor seal Algunos materiales de rotor pueden adsorber menos la muestra que otros por lo que puede merecer la pena investigar si el efecto se reduce o se elimina cambiando entre Vespel, Tefzel y PEEK. El lavado regular con fase móvil puede ayudar a minimizar la contaminación del rotor seal. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 78 - Incidencias y Mantenimiento en HPLC Ajuste del volumen de inyección al poder de elución del disolvente de la muestra En condiciones ideales el poder de elución del disolvente de la muestra no debería afectar a la separación cromatográfica, ya que éste se diluiría de forma instantánea hasta tener la misma composición que la fase móvil. Sin embargo, esta dilución del disolvente de la muestra lleva en la práctica un periodo de tiempo finito para que la fase móvil la realice. Las siguientes figuras ilustran el deterioro de la forma de pico debido a una inadecuada combinación de disolvente de muestra y fase móvil, mostrando la inyección de una mezcla test de cuatro componentes disueltos en diferentes disolventes y separados en una columna C18 de fase reversa en condiciones isocráticas de MeOH:H2O 60:40 (v/v). En cada cromatograma la mezcla test estaba disuelta en: • • • • (a) − Fase Móvil (b) − Tetrahidrofurano (c) − Etanol (d) − Butanol La siguiente tabla proporciona unas guías para elegir el volumen de inyección en función del poder de elución de los disolventes de la muestra con respecto a la fase móvil. Fuerza Eluotrópica del Disolvente Volumen Máximo de Inyección 100% Disolvente Fuerte ≤10µL Algo más fuerte que la Fase Móvil ≤25µL Fase Móvil ≤15% del volumen de pico Más débil que la Fase Móvil Grande Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 79 - Incidencias y Mantenimiento en HPLC 100% Disolvente Fuerte En esta exigente situación es necesario mantener el volumen de inyección lo más bajo posible para evitar la distorsión de los picos. La recomendación es no superar los 10µL. Cuando el disolvente de la muestra es más fuerte que el 80% de Fase Móvil se pueden inyectar volúmenes mayores puesto que la diferencia de composición entre el disolvente de la muestra y la fase móvil es proporcionalmente menor. Más Fuerte que la Fase Móvil Cuando el disolvente de la muestra no es más de un 25% más fuerte que la fase móvil, es posible inyectar volúmenes de hasta 25µL. Observe no obstante el cromatograma para vigilar la posible distorsión en los picos dada la interrelación existente entre esta regla y la anterior. En general, los problemas de solubilidad de los analitos son la razón principal para aumentar el poder de elución de los disolventes de muestra empleados. Para minimizar las variaciones de poder elución en la inyección, se recomienda disolver el analitos en una alta concentración del disolvente de mayor poder de elución y después, diluirlo con el disolvente más débil para corregir progresivamente la diferencia de fuerza eluotrópica. En el peor escenario posible hay que disolver la muestra en un 100% de disolvente fuerte y mantener el volumen de inyección al mínimo posible. Fase Móvil La mejor situación posible es que el disolvente de la muestra y la fase móvil sean iguales, tanto en poder de elución como en pH. En esta situación, debido a la homogeneidad de disolventes, no hay que preocuparse por los efectos de dilución. Sin embargo, el volumen de inyección es limitado. La contribución del volumen de inyección al ensanchamiento del pico cromatográfico puede determinarse con la siguiente ecuación: Vol. Muestra.Inyectada2 + Vol.PicoColumna2 = Vol.PicoTotal2 Si el pico más estrecho (p.ej. el primero) fuese de 30 de anchura a línea de base, Su volumen de pico en columna (Vol.PicoColumna) a un flujo de 1mL/min sería igual a, (1mL/min / 60) x 30 = 500µL En consecuencia, el volumen máximo de muestra que puede inyectarse sin aumentar el ancho de pico más de un 1% sería igual a, Vol. Vol. Vol. Vol. Vol. Muestra.Inyectada2 = Vol.PicoTotal2- Vol.PicoColumna2 Muestra.Inyectada2 = (505µL)2 – (500µL)2 Muestra.Inyectada2 = 5025 Muestra.Inyectada = √5025 Muestra.Inyectada = ~70µL Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 80 - Incidencias y Mantenimiento en HPLC Más Débil que la Fase Móvil Para establecer el efecto del poder de elución de la fase móvil hay que considerar la “Regla de Tres”, que establece que un cambio del 10% en el disolvente fuerte producirá aproximadamente un cambio de tres veces en el tiempo de retención de los analitos. Por lo tanto, si un pico cromatográfico tiene un tiempo de retención de 5min en una fase móvil 40:60 agua:acetonitrilo, en una fase móvil de 60:40 agua:acetonitrilo su tiempo de retención aumentará hasta aproximadamente 45min (3 x 3 x 5min = 45min). En consecuencia, si la muestra se inyecta en 60:40 agua:acetonitrilo, las moléculas de analito viajarán más lentamente a través de la columna, hasta que el disolvente de muestra sea completamente diluido por la fase móvil. Las bandas cromatográficas que se mueven más lentamente que la fase móvil tienden a comprimirse, debido a que la fase móvil alcanza a las moléculas de analitos en la “cola” del pico tenderán a moverse más rápido en la columna, cazando a las moléculas de analito adelantadas que se encuentran en la fase móvil de menor poder de elución. Cuando la diferencia entre la fuerza eluotrópica del disolvente de la muestra y la de la fase móvil aumenta, es posible aumentar progresivamente cada vez más el volumen de inyección. Este aumento permite un “enfoque”, o “concentración” de los analitos en la columna y se emplea a menudo en análisis medioambientales para facilitar la detección de trazas de pesticidas. Excepciones a lo anterior son los casos en que la fase móvil contenga aditivos a nivel de traza, en ese caso se recomienda siempre inyectar las muestras disueltas en fase móvil. P.ej. las separaciones por par-iónico se basan en el equilibrio entre el reactivo de par-iónico entre la fase móvil y la estacionaria. Si se inyecta la muestra en un disolvente que no contenga el par-iónico este equilibrio se desplaza a la fase móvil provocando distorsión en los picos. Aunque las inyecciones de loop parcial son generalmente menos precisas que las que realizan el llenado completo del loop, este problema se reduce con los automuestreadores gracias a que su elevada precisión en la inyección hace que este error sea constante. En condiciones normales la línea de desechos (“waste”) de los a automuestreadores de loop integrado nunca se utiliza. Pueden aparecer problemas de precision cuando la línea de desechos se obstruye parcialmente con sales de los tampones o cualquier otro residuo de la evaporación del disolvente de la muestra. Se recomienda lavar la línea de desechos regularmente, lo que será una práctica habitual de la operación con automuestreadores. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 81 - Incidencias y Mantenimiento en HPLC Debe considerarse la viscosidad del disolvente de la muestra. Si la aguja de inyección intenta extraer a demasiada velocidad el volumen de un disolvente viscoso, puede que el volumen de inyección seleccionado no entre completamente en el loop. Este fenómeno se denomina “cavitación”, y produce formación de burbujas, comprometiendo la repetibilidad del automuestreador. Pueden aparecer también problemas de precisión si el vial de muestra se llena en exceso. En estas circunstancias se produce un vacío parcial, que es suficiente para producir cavitación cuando se extrae la muestra. Se recomienda no llenar los viales a más de tres cuartos de su capacidad para minimizar el impacto de dichos efectos. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 82 - Incidencias y Mantenimiento en HPLC Problemas Mecánicos Una operación fiable del automuestreador requiere que se mantenga limpio y se compruebe regularmente su adecuado ajuste. Hay que limpiar rápidamente todas las salpicaduras de tampones y/o reactivos para prevenir la creación depósitos, que pueden corroer o restringir el movimiento del automuestreador. Por esta razón los viales de muestra nunca deben rellenarse cerca del autoinyector. Debe comprobarse periódicamente el alineamiento de la gradilla de muestras, muchos fabricantes incorporan mensajes de error en sus softwares para avisar que la gradilla no está o está incorrectamente colocada. Muy importante, el automuestreador es una parte del sistema HPLC que debe lavarse exhaustivamente con fase móvil después de análisis de muestras con tampones o fases móviles tamponadas. Reemplazar en primer lugar el componente tampón de la fase móvil por H2O para eliminar cualquier depósito de sales potencialmente corrosivas en la válvula de inyección, aguja del inyector y capilares. Después, extrayendo la columna si fuera necesario, lavar con H2O para asegurar que todas las trazas de las sales del los tampones se eliminan completamente. Posición de la Gradilla e Identificación de Muestra Las muestras incorrectamente identificadas pueden provocar potencialmente consecuencias graves en los resultados reportados. Aunque las imprecisiones de la inyección se pueden corregir con un apropiado estándar interno y un bloqueo o fallo mecánico se evidencia porque no se realiza la inyección de la muestra, el posicionamiento del automuestreador es crucial para una inyección correcta. Los automuestreadores emplean sensores ópticos o mecánicos para asegurar la adecuada selección del vial, y éstos deben mantenerse limpios y en adecuado funcionamiento. Una rápida revisión visual de los cromatogramas y los datos de muestra asociados para comprobar si los estándares se correlacionan adecuadamente, antes de sacar ningún vial del automuestreador, permitirá la identificación de cualquier muestra que requiera un re-análisis. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 83 - Incidencias y Mantenimiento en HPLC Efecto Memoria El Efecto Memoria aparece cuando la inyección de un blanco de disolvente produce una mini versión del cromatograma inyectado previamente. En la mayoría de los casos este efecto se produce en la válvula de inyección debido a adsorción de muestra. El Efecto Memoria entre inyecciones puede eliminarse de diferentes formas: • Implementando un régimen de lavado de la aguja entre inyecciones. Los automuestreadores de loop integrado están configurados para mantener la orientación de la inyección de muestra, permitiendo así el lavado continuo del sistema de inyección y de la válvula de inyección durante todo el cromatograma y reduciendo o eliminando de esta forma el efecto memoria. Otros diseños de automuestreadores permiten lavados de aguja y/o de jeringa entre las inyecciones. Esta solución de lavado puede provenir de una fuente externa rellenada independientemente, o bien puede encontrarse en la propia gradilla del automuestreador en una posición definida por el usuario. • Minimizando el impacto del Efecto Memoria La dilución de muestra, además de mejorar la precisión del automuestreador como se estudió anteriormente, reduce el efecto memoria porque los automuestreadores tienen un volumen muerto finito p.ej. 0.1µL, que representa el volumen de muestra residual. Por lo tanto, con una inyección de muestra de 10µL este volumen residual generaría un 1% de efecto memoria, pero sería solo del 0.1% para una inyección de 100µL. Como alternativa a la dilución se recomienda ordenar las muestras de menor a mayor concentración, ya que un efecto memoria de volumen fijo generará menos problemas cuando a una concentración baja le sigue una mayor que al revés. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 84 - Incidencias y Mantenimiento en HPLC El Efecto memoria, junto a una mala precisión en áreas y alturas, son indicios de un rotor seal agotado. El rotor seal debe revisarse para buscar síntomas de desgaste, especialmente un ahondamiento en los microcanales entre los puertos que puede generar puntos de retención de muestra localizados. Estos puntos se “limpian” en la siguiente inyección produciendo un mini-cromatograma o “picos fantasma”. El Efecto memoria o los picos fantasma también pueden ser consecuencia de materiales adsorbidos provenientes de la inyección previa que han sido desorbidos en la nueva solución de muestra inyectada. Tal adsorción puede ocurrir en varios puntos del sistema de inyección, y puede comprobarse lavando éste con 10 x 5 volúmenes de loop para a continuación re-inyectar un blanco. Si no aparecen picos hay que optimizar la rutina de lavado o investigar posibles modos de adsorción de los componentes de la muestra en la superficie de los componentes del inyector y corregirlos cuando sea necesario. Algunos materiales de rotor pueden adsorber menos la muestra que otros por lo que puede merecer la pena investigar si el efecto se reduce o se elimina cambiando entre Vespel, Tefzel y PEEK. Fugas El Aire puede introducirse potencialmente en el sistema a través de las paredes permeables de los tubos de PTFE empleados en el automuestreador. Esto no representa problemas si se utilizan en su lugar tubos de acero inoxidable o PEEK en la sección de toma de muestra de la unidad. Una tuerca conectada o parcialmente apretada en la conexión del loop de muestra con la aguja de inyección puede también provocar la entrada de aire cuando la muestra se succiona desde el vial. Un cuerpo de la válvula de inyección mal montado, o un componente “sospechoso” pueden producir también fugas en el cuerpo de la válvula. Desmonte la válvula y vuelva a montarla según las instrucciones del fabricante para solucionarlo. Debido a su creciente complejidad, los inyectores más caros tienen más áreas con fallos potenciales, y por lo tanto, mayor mantenimiento. Sin embargo, también disponen de sistemas mejorados de detección de fugas y/o burbujas y una mayor precisión en la inyección de volumen variable. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 85 - Incidencias y Mantenimiento en HPLC 6 Columnas Los objetivos de esta sección son: • • Describir los procesos generales de limpieza, purga y almacenamiento de las columnas HPLC. Relacionar y describir los procedimientos de diagnóstico y mantenimiento más comunes. 6.1 Limpieza, Purga y Almacenamiento de la Columna Limpieza Las columnas deben limpiarse antes de su purga y almacenamiento ya que este proceso permite regenerar la superficie de la fase estacionaria después de un uso prolongado, asegurando así la eliminación de contaminantes o aditivos de la fase móvil que pudieran haber quedado fuertemente retenidos. Este proceso tiene la misma importancia tanto si la columna se ha utilizado para una única aplicación o analito en particular, como si ha sido empleada para la separación de analitos o métodos diferentes. La limpieza de columna debe realizarse rutinariamente al final de cada lote de muestras cuando se emplea una separación isocrática para prevenir de esta forma la acumulación de contaminantes provenientes de las muestras. Los métodos que empleen análisis en gradiente en cambio, limpian ya la columna cuando la proporción del disolvente de mayor poder de elución se aumenta al final de cada análisis. Limpiar exhaustivamente una columna nos asegura que el siguiente usuario no experimentará tiempos de equilibrio prolongados ni deberá realizar un gran número de inyecciones de acondicionamiento. Purga de la Columna Después de la Limpieza, la columna debe ser convenientemente purgada para eliminar cualquier traza de sales (tampones) y/o aditivos provenientes de la fase móvil. Estas sales pueden precipitar introduciendo partículas que causarán bloqueos provocando el aumento de la presión de trabajo. En ocasiones, algunas sales empleadas como tampones, pueden ser de naturaleza corrosiva pudiendo provocar daños en la superficie metálica del interior de la columna si quedasen retenidas en el ella varios días, generando así un aumento del ruido de línea de base o incluso produciendo fugas. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 86 - Incidencias y Mantenimiento en HPLC Almacenamiento de la Columna La lectura de la documentación proporcionada por el fabricante de la columna nos indicará cuál es el disolvente apropiado para su almacenamiento. Si se requiere cambiar de disolvente a otro no miscible con el que actualmente contiene la columna, deberá emplearse un disolvente intermedio que sea miscible con ambos. Al desconectar la columna del equipo deben apretarse convenientemente los tapones para evitar la evaporación del disolvente de almacenamiento y el consiguiente secado de la fase estacionaria. A continuación se almacenamiento que estacionarias con base del fabricante para columna. describe un protocolo genérico de purga y puede utilizarse con la mayoría de las fases de sílice. Por favor, consulte la literatura específica comprobar requerimientos especiales para cada 1. Sustituya la fase móvil que contiene el tampón por H2O, y cambie a una composición idéntica de H2O: orgánico a la utilizada en el método de separación original. Esta etapa limpia de forma muy eficiente la columna y el sistema dejándolos libres de cualquier sal de los tampones que pudiera precipitar. Por ejemplo, Si la fase móvil es 50mM KH2PO4 y Acetonitrilo 60:40 (v/v), sustituiremos el 60% de tampón KH2PO4 con Agua purgando abundantemente. Se recomienda utilizar típicamente 10 volúmenes de columna, aunque si se han empleado reactivos formadores de pares de iones, pueden ser necesarios de 30 a 50 volúmenes para asegurar la limpieza correcta. 2. Después de la purga pasar a 95:5 (v/v) H2O: orgánico. Este proceso puede realizarse a una velocidad de cambio del 10% de orgánico por minuto, de forma que el alto contenido de H2O elimine todas las trazas de sales tampón, eliminado así la posibilidad de precipitados posteriores al pasar eluyentes que contengan una alta concentración de orgánico. Se recomienda en esta etapa purgar con 5 volúmenes de columna. Nunca se deben usar las columnas de fase reversa a un 100% H2O o podrían quedar desactivadas por un colapso de fase. El colapso de fase se produce habitualmente cuando la fase móvil tiene un contenido de agua mayor del 95%, y se describe como una auto-asociación de los ligandos hidrofóbicos alquil y aril de la fase estacionaria en un intento de evitar a un eluyente altamente polar. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 87 - Incidencias y Mantenimiento en HPLC Como consecuencia de este proceso la superficie de la sílice se desactiva con respecto a los analitos no polares y sus tiempos de retención se reducen con la consiguiente pérdida de resolución. La Figura muestra la auto-asociación de los ligandos de la fase estacionaria en presencia de una fase móvil altamente acuosa. Solo debe emplearse un 100% H2O si la columna ha sido diseñada por el fabricante como compatible con fases móviles 100% acuosas. Las columnas que son capaces de trabajar a 100% de agua tienen típicamente una fase estacionaria modificada en la que los ligandos se han protegido del colapso de fase incorporándoles un grupo funcional polar en el interior de su estructura. La misión de ese grupo funcional es adsorber una capa de H2O de forma que se sitúe bajo la porción hidrofóbica del ligando eliminándose así la posibilidad de un colapso de fase. La Figura muestra la activación de la fase estacionaria en presencia de un solvente orgánico. 3. Pase a 100% de solvente orgánico y lave el sistema y la columna exhaustivamente para eliminar los contaminantes, incluyendo aquellos fuertemente absorbidos provenientes de la matriz de las muestras. Este proceso puede realizarse a 10% orgánico por minuto. Se recomiendan típicamente cambios de 5 volúmenes de columna. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 88 - Incidencias y Mantenimiento en HPLC 4. Cambie la composición a 40:60 (v/v) H2O:orgánico como disolvente genérico de almacenamiento para columnas HPLC con base de sílice. Esta composición de la fase móvil evita el riesgo de crecimientos microbianos, mantiene la superficie de la sílice “empapada” y evita la formación de células electrolíticas que podrían crear depósitos de iones metálicos provenientes de las fritas en la sílice del relleno, provocando la activación de silanoles residuales e introduciendo efectos de columna secundarios. Se recomienda utilizar típicamente 5 volúmenes de columna. Este protocolo también es adecuado para el almacenamiento del instrumento. A continuación se muestra de forma esquemática el proceso de purga y almacenamiento. Si la presión de trabajo del sistema lo permite, el flujo puede aumentarse a 2 mL/min para realizar un cambio más rápido en la composición H2O:organico. El protocolo completo puede programarse en un método de purga, que puede ejecutarse al final de cada secuencia o lote de muestras. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 89 - Incidencias y Mantenimiento en HPLC 6.2 Incidencias de la Columna El diagnóstico de las columnas se realiza habitualmente a partir del estudio del cromatograma y de los picos cromatográficos. Los parámetros de operación del sistema también proporcionan información útil para el diagnóstico (la perspectiva de los componentes). Siempre es recomendable proteger a las columnas de deterioro debido a la obstrucción por partículas o muestras fuertemente retenidas. Éstas pueden bloquear eventualmente la frita de entrada u ocluir la fase estacionaria provocando un aumento en la presión de trabajo o una pobre forma del pico cromatográfico respectivamente. Es muy recomendable por tanto la instalación de filtros de línea y/o guardacolumnas en el sistema. Las partículas y las especias fuertemente adsorbidas pueden provenir potencialmente de: • Sellos de los pistones • Sello del rotor de la válvula de inyección. • Sales de los tampones precipitadas o no disueltas. • Matrices de las muestras contaminadas. La tabla siguiente relaciona los problemas más frecuentes con las columnas: Problema con la columna % de frecuencia Problemas con la presión 24 Mala reproducibilidad 16 Recuperación de la muestra 14 Pérdida de resolución 13 Inestabilidad 11 Formación de volúmenes muertos 8.1 Fugas en las conexiones 4.8 Rango de pH 2.0 Baja eficacia 2.0 Saturación de las columnas 2.0 Costes 1.7 Otros 1.5 Fuente − LC-GC Europe (1994) Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 90 - Incidencias y Mantenimiento en HPLC Uso de las columnas Una vez se ha usado una columna para la separación de muestras “reales” la selectividad proporcionada ésta variará necesariamente de la ofrecida por una columna nueva de la misma marca y referencia. Las Columnas pueden llegar a modificarse de diversas formas una vez se utilizan de forma experimental: • • • En condiciones de pH bajo, la columna puede perder las especies activas o ligandos (endcapping). Algunos materiales fuertemente retenidos pueden enlazarse de forma irreversible a la cabeza de columna provocando un aumento de la presión del sistema. Los analitos de naturaleza fuertemente básica pueden quedar enlazados irreversiblemente a los grupos silanol libres de la fase estacionaria. Este ionización de los silanoles puede reducirse realizando las separaciones a bajo pH, ya que habitualmente éstos grupos muestran valores de pka entre 3.8−4.1. Puesto que cada método HPLC opera en diferentes condiciones analíticas y se utiliza para diferentes tipos de muestras, los cambios que potencialmente puedan producirse en la columna serán únicos para cada método. Por lo tanto, una vez que una columna haya sido usada para una determinada aplicación, puede no volver a ofrecer nunca una separación idéntica a la obtenida en una columna equivalente nueva, observándose a menudo importantes diferencias cromatográficas entre ellas. Este hecho también puede observarse cuando se utiliza una misma columna con diferentes métodos y analitos. En este caso pudiera ser necesario realizar varias inyecciones de acondicionamiento (“priming” injections) con el nuevo analito para llegar a conseguir una cromatografía reproducible. Los tiempos de equilibrado de la columna deberán a menudo aumentarse para adecuar la columna a los cambios en la selectividad. Con el fin de evitar en lo posible la aparición de tales variaciones, se recomienda que cada columna se dedique a un método y tipo de analítico específico. El intercambio indiscriminado de columnas para el desarrollo de métodos puede producir una separación cromatográfica no representativa una vez se traslade a una columna nueva, que no habría sido expuesta a la variedad de muestras de su predecesora. Estas variaciones pueden reducirse si se emplea un procedimiento adecuado de lavado de la columna. Filtros de Línea Una de las causas más comunes del deterioro de la columna es el aumento excesivo de la presión de trabajo. Este aumento de la presión se produce habitualmente por la acumulación de material particulado en la frita de entrada a la columna, y una de las formas más económicas y efectivas de aumentar la vida de ésta es incorporar un filtro de línea de alta presión Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 91 - Incidencias y Mantenimiento en HPLC entre el Automuestreador y la Columna. Un filtro de línea situado en esa posición evita que las partículas provenientes de la muestra o del rotor de la válvula de inyección del automuestreador alcancen la cabeza de la columna. Es MUY recomendable incluir un filtro de línea de alta presión en todos los sistemas HPLC con la única excepción de los sistemas de volumen muerto extremadamente pequeño, en los que el incremento “extra” en el volumen de columna puede contribuir al ensanchamiento y dispersión de los picos. Fuera de este escenario no se observarán cambios apreciables en el cromatograma. El filtro de línea puede examinarse visualmente para observar si hay depósitos de contaminación o cambios en el color de su superficie cuando la presión del sistema aumenta apreciablemente. El tamaño más habitual de poros para estos filtros está alrededor de 0.5µm. Limpio − el filtro puede reinsertarse para su uso. Sucio –El filtro ha cambiado de color o muestra depósitos en su superficie. Descartar y reemplazar. Figura mostrando filtros de línea limpio y contaminado. Guarda Columnas/Discos Una guarda columna es una columna corta – típicamente de 10-20 mm de longitud- que contiene la misma fase estacionaria que la columna analítica. Si se utiliza un sistema de cartuchos, entonces es necesario utilizar un soporte de cartuchos adecuado. Los discos guarda columna son pequeños discos fritados que cuando se alojan en su soporte se enroscan directamente en la columna analítica. En comparación con un filtro de línea, una guarda columna o disco debe utilizarse solamente como filtro “químico”, para asegurar la eliminación de cualquier material potencialmente retenido o agresivo evitando así el posterior fallo posterior de la columna analítica. En algunos casos una guardacolumna puede proporcionar suficiente protección como para eliminar el “clean up” (preparación de muestra previo) de la muestra. Sin embargo, ya que una guarda columna puede quedar contaminada con una gran variedad de materiales fuertemente retenidos, es un requisito obligatorio que cuando se lave con un disolvente fuerte se haga sin conexión con la columna analítica, evitando así que las Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 92 - Incidencias y Mantenimiento en HPLC especies peligrosas pasen inadvertidamente a la cabeza de la columna analítica. Nunca debe usarse una guarda columna como columna de saturación de sílice, ya que cualquier volumen muerto generado en la guarda columna comprometerá necesariamente la cromatografía. Si la columna saturadora se estima necesaria para una aplicación en particular (p.e. Fase Móvil de alto pH), debe ser instalada antes del inyector y con un filtro de línea inmediatamente después para retener las pequeñas partículas de sílice resultantes de la disolución de la sílice. La Figura muestra el efecto negativo de formación de un volumen muerto en la guardacolumna con el consiguiente deterioro de la forma de pico. Tanto la incorporación al sistema de HPLC de un filtro de línea como la de una guarda columna, provocan una aumento del volumen extra-columna, que podría contribuir a un mayor ensanchamiento de pico en sistemas con un volumen muerto extremadamente pequeño. Figura mostrando guarda columnas y cartuchos típicos. Figura mostrando un disco guarda típico. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 93 - Incidencias y Mantenimiento en HPLC ¿Cuándo se debe sustituir una guarda columna? Existen tres procedimientos fundamentales para determinar cuándo sustituir una guarda columna: 1. ¡No se recomienda esperar al deterioro de la separación cromatográfica! Si es posible, se puede monitorizar la separación de una pareja de picos del cromatograma que estén peor resueltos que los picos de interés, estableciendo un criterio para predecir el momento adecuado antes de que la guarda columna potencialmente falle. Esto se muestra en el cromatograma de abajo, que muestra la separación de un grupo de herbicidas. La pareja crítica, los picos cromatográficos que están peor resueltos uno del otro, están marcados como 1 y 2. Los picos cromatográficos de interés están marcados como 3 y 4. Si monitorizamos la separación entre los picos 1 y 2 a lo largo del tiempo, puede establecerse un criterio de valoración que nos permita cambiar la guarda columna sin comprometer el análisis con respecto a los picos de interés 3 y 4. 2. 3. Monitorizar el número de muestras inyectadas con la guarda columna instalada hasta que el sistema deje de proporcionar una separación satisfactoria. Modificar el PNT de forma que la guarda columna se sustituya aproximadamente al 80% del tiempo de vida esperado – ¡sea proactivo en lugar de reactivo! Monitorizar la vida efectiva de la columna en función del volumen de eluyente empleado o de la fecha en la que la guarda columna fue instalada. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 94 - Incidencias y Mantenimiento en HPLC Limpieza (Clean-up) de la muestra En el contexto de la limpieza de muestra siempre existe un balance entre los costes del citado proceso de limpieza frente a los costes de sustitución de la columna. Consideremos un ejemplo bioanalítico en el que se desea medir la concentración de un fármaco en plasma. La precipitación de las proteínas por medio de la adición de acetonitrilo al plasma, agitación en Vortex, centrifugación e inyección del sobrenadante producirán muestras “razonablemente” sucias permitiéndonos quizás entre 100 y 500 inyecciones. Por el contrario, una muestra mucho más limpia obtenida tras un procedimiento de limpieza por Extracción en Fase Sólida (SPE), nos permitirá obtener una vida media de unas 1000-2000 inyecciones. Este proceso SPE y posterior inyección de muestra puede automatizarse empleando un formato de 96-pocillos. Mientras que la ganancia en tiempo de vida de la columna pudiera no justificar por sí sola el coste de este elaborado y caro proceso de limpieza, el método se beneficiaría de la mejora en reproducibilidad lo que considerado a lo largo de los años debería tenerse en cuenta. Sustitución de la Columna Poniendo en práctica las recomendaciones detalladas hasta ahora podremos incrementar apreciablemente la vida de las columnas pero en definitiva éstas deben considerarse como materiales consumibles. La duración de una columna viene determinado por un gran número de factores interrelacionados, p.ej. Tampones y su pH, limpieza de muestra, etc, pero habitualmente un rango entre 500-2000 inyecciones puede considerarse aceptable. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 95 - Incidencias y Mantenimiento en HPLC 7. Picos con Cola La aparición de colas en los picos es una de las deformaciones cromatográficas más frecuentes. Se dice que un pico tiene cola cuando muestra una asimetría mayor de 1.2, aunque en ocasiones picos con valores de As tan altos como 1.5 puedan ser aceptables para una gran variedad de ensayos. El factor de asimetría de pico (π) se calcula según la siguiente ecuación: π = wb2 / wb1 Donde; wb2 = Ancho del pico en la línea de base después del centro del pico wb1 = Ancho del pico en la línea de base antes del centro del pico La principal causa de aparición de cola en un pico es la coincidencia de más de un mecanismo de retención. En las separaciones por Fase Reversa, el principal modo de retención de los analitos es a través de interacciones hidrofóbicas no-específicas con la fase estacionaria. Sin embargo, también son frecuentes las interacciones polares con grupos de silanoles residuales existentes en el soporte de sílice. Compuestos que posean en su estructura grupos amino o cualquier otro grupo básico, pueden interactuar fuertemente con los grupos silanoles ionizados, produciendo picos con cola. Este efecto se muestra en la figura siguiente a un pH >3.0. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 96 - Incidencias y Mantenimiento en HPLC Estas interacciones secundarias pueden minimizarse si se realiza la separación a un pH más bajo, asegurando así la protonación completa de los citados grupos silanol residuales; Otra alternativa es la utilización de columnas “end-capped”. “End-capping” es el nombre que se da al proceso químico por el cual los grupos silanoles residuales de la fase estacionaria se transforman en otros grupos funcionales sustancialmente menos polares en su superficie. Esta modificación tiene el efecto de reducir las interacciones secundarias que puedan producirse potencialmente con moléculas de analitos polares. Los reactivos químicos empleados para el proceso de “end-capping” incluyen generalmente; • • Trimetilclorosilosano (TMCS) Hexametildisilazano (HMDS) Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 97 - Incidencias y Mantenimiento en HPLC El proceso de “End-capping” reduce las colas observadas con analitos polares, bloqueando de forma eficiente sus interacciones con los grupos silanol residuales que pudieran ser potencialmente ionizables. A pesar de ello, el “end-capping” sólo disminuye aproximadamente a un 50% el número de grupos silanol no reaccionados. Debido a factores de impedimento estérico este tipo de reacciones rara vez son eficientes en términos absolutos y en consecuencia una columna completamente “end capped” no es nunca tanto como debería! En el caso de que todos los picos cromatográficos muestren cola debe considerarse la posibilidad de que la columna se haya sobrecargado por un exceso de muestra. Evalúe aplicando la ecuación descrita previamente en la página 104. Si fuera necesario utilice una fase estacionaria de mayor capacidad p.ej. de mayor % carbono o tamaño de poro, utilice una columna de mayor diámetro, o reduzca la cantidad absoluta de muestra o volumen inyectado. Examine la columna por si se hubieran formado volúmenes muertos o se hubiera bloqueado parcialmente la frita de entrada. Sustituir la columna por otra nos confirmará rápidamente el problema. Si se sospecha que existe algún volumen muerto, quite las conexiones de cierre y revise la cabeza de columna. Si se observa algún hueco en el relleno cúbralo con fase estacionaria, o si no, limpie o sustituya la frita de entrada y pruebe la columna de nuevo para ver si el problema estaba originado o no por una frita parcialmente obstruida. En ocasiones, invertir la columna y lavarla con un 100% de un disolvente fuerte también permite eliminar bloqueos y contaminación del filtro de entrada. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 98 - Incidencias y Mantenimiento en HPLC Compruebe las instrucciones del fabricante para ver si la columna puede mantenerse en la orientación inversa de flujo o no. Si sólo uno, o algunos, de los picos cromatográficos muestran cola, considere la posibilidad de que la columna esté mostrando efectos de partición o retención secundarios. Reduzca la probabilidad de que cualquier analito potencialmente básico interactué con los grupos silanol residuales usando columnas fabricadas con sílice desactivada (Tipo B), que han sido completamente bloqueadas (“endcapped”). Como parte de cualquier procedimiento de desarrollo de métodos reduzca el pH de la fase móvil para suprimir la ionización de cualquier grupo silanol. En general, una fase móvil con pH <3 será suficiente para este propósito. Adicionalmente, el empleo de fases móviles a bajo pH asegura la protonación de los analitos ácidos, aumentando su retención y minimizando cualquier tipo de interacción iónica. La figura de abajo ilustra el efecto producido en las colas de pico al reducir el pH de separación. La mezcla de cinco fármacos básicos consiste en Fenilpropanolamina, Efedrina, Anfetamina, Metanfetamina y Fenteramina (Picos 1−5) respectivamente. El primer cromatograma se adquirió a un pH de la fase móvil de 7.0, y en estas condiciones de separación, el Pico 4, Metanfetamina muestra un factor de cola USP al 5% (USP Tf 5%) de 2.35. La disminución del pH de la fase móvil reduce la interacción de los analitos básicos con los grupos silanol residuales eliminado de esta forma la cola excesiva de los picos. El segundo cromatograma se adquirió con una fase móvil a pH 3.0 y el USP Tf (5%) de la Metanfetamina en estas condiciones se redujo a 1.33. La adición de Trietilamina (TEA) a la fase móvil en niveles de concentración de 5−10mM, también reduce o posiblemente elimina, la cola en compuestos básicos. La TEA compite con los analitos polares básicos por enlazarse con los grupos silanol residuales, produciendo una función de “pseudo endcapping”. Esta alternativa sin embargo es menos conveniente, ya que la fase estacionaria se altera irreversiblemente por la adición de tal Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 99 - Incidencias y Mantenimiento en HPLC modificador, afectando a la selectividad mostrada por el sistema de separación posteriormente. La cromatografía de pares de iones puede resolver el problema de las colas para analitos ácidos o básicos. Otra opción para la separación de especies iónicas podría ser emplear cromatografía de intercambio iónico. Desde un punto de visto práctico, generalmente sólo es posible utilizar el pH para suprimir la ionización ácida ya que la ionización básica requiere valor de pH típicamente mayores de 8, y a ese pH se puede producir la disolución de la sílice. Sin embargo, los fabricantes de columnas ofrecen versiones que permiten trabajar a un pH mayor, protegiendo la sílice de su disolución por medio de ligandos bi- y tri- dentados. Estos ligandos son en realidad dos o tres ligandos tradicionales que se han puenteado por medio de procesos químicos patentados antes de aplicarlos a la fase estacionaria. Su estructura puenteada permite proteger su superficie frente a valores extremos de pH. La Figura muestra ligandos bidentados y la protección ofrecida a la superficie de la sílice gracias a su estructura de puente. Un único pico con cola también podría producirse por un pequeño pico eluyendo inmediatamente después del pico cromatográfico de interés. La posibilidad de que esto ocurra es la razón primordial por la que las colas de pico nunca deben ignorarse. Como se describía en el apartado “desdoblamiento de picos y hombros”, podemos confirmar la presencia de un interferente cambiando la longitud de onda de detección, o mejorar la resolución de la separación empleando una columna de mayor eficiencia p.ej. una columna más larga o empaquetada con partículas de menor tamaño. Ajuste la selectividad del sistema frente a compuestos co-eluyentes variando la composición o el tipo de la fase móvil, la temperatura, o la fase estacionaria de la columna. Emplee algún procedimiento de “clean-up” p.ej. SPE para eliminar cualquier contaminante interferente. Evalúe si alguno de los picos coeluyentes pueden provenir de inyecciones anteriores haciendo una inyección en blanco y siguiendo el procedimiento descrito en “Ensanchamiento de Picos” de la pág. 105. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 100 - Incidencias y Mantenimiento en HPLC 8. Picos con Frente Si todos los picos cromatográficos muestran frente debe considerarse la posibilidad de una sobrecarga de la columna por exceso de muestra inyectada. Evalúelo aplicando la ecuación descrita previamente en la pág. 104. Si fuera necesario utilice una fase estacionaria de mayor capacidad p.ej. de mayor % carbono o tamaño de poro, utilice una columna de mayor diámetro, o reduzca la cantidad absoluta de muestra o volumen inyectado. Si se ha inyectado un volumen de muestra excesivamente grande, y el solvente de la muestra y la fase móvil no están adecuadamente emparejados, pueden producirse dos equilibrios de distribución, provocando una partición diferencial en la columna. Refiérase a las reglas empíricas descritas en las páginas 54-56 en relación al adecuado emparejamiento de entre el volumen de inyección con la fuerza eluotrópica del disolvente de la muestra para eliminar éste fenómeno. Un único pico que muestre frente también puede deberse a un pequeño pico que eluya justo antes del pico cromatográfico de interés. Esta posibilidad es la principal razón por la que un pico con frente nunca debe ser ignorado. Como se describía en el apartado “desdoblamiento de picos y hombros”, podemos confirmar la presencia de un interferente cambiando la longitud de onda de detección, o mejorar la resolución de la separación empleando una columna de mayor eficiencia p.ej. una columna más larga o empaquetada con partículas de menor tamaño. Ajuste la selectividad del sistema frente a compuestos co-eluyentes variando la composición o el tipo de la fase móvil, la temperatura, o la fase estacionaria de la columna. Emplee algún procedimiento de “clean-up” p.ej. SPE para eliminar cualquier contaminante interferente. Evalúe si alguno de los picos coeluyentes pueden provenir de inyecciones anteriores haciendo una inyección en blanco y siguiendo el procedimiento descrito en la pág. 89. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 101 - Incidencias y Mantenimiento en HPLC Tutorial 2 Pregunta 1 Usando la tabla siguiente enumere cinco partes del Automuestreador de Loop completo que pueden requerir mantenimiento rutinario. Indique la principal incidencia que puede presentar cada una de los componentes enumerados y cuál sería la solución más adecuada para remediarla. Componente Incidencia Traducción Grupo Biomaster 2009 Solución © Crawford Scientific 2007 - 102 - Incidencias y Mantenimiento en HPLC Pregunta 2 A continuación se muestran dos cromatogramas que pueden o no darnos información sintomática del status operacional del sistema HPLC. Indique que acciones pueden ser necesarias para mejorar la cromatografía obtenida. Rellene sus respuestas empleando la tabla incluida. Condiciones: Muestra: Columna: Flujo: Fase Móvil: Vol Inyección: Detección: Cuatro Esteroides Anabolizantes de un suero concentrado. Concentración estimada − 10ug/mL. RP C18 columna 150 x 4.6 mm 5µm Obtenida sin ninguna información histórica previa. 1.5mL/min 46% Agua- MeOH:0.01% KHPO4 v/v (pH 5.2) 100µL 254nm Incidencia Solución Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 103 - Incidencias y Mantenimiento en HPLC 9 Detectores El objetivo de esta sección es: • • • • Describir las propiedades del detector ideal. Definir los términos y conceptos más comunes. Describir el diseño y los principios de operación de los detectores de HPLC más comunes. Ennumerar y describir las incidencias más importantes con los detectores y los procedimientos de mantenimiento. 9.1 El Detector Ideal El Detector ideal debería mostrar las siguientes características: 1. 2. 3. 4. 5. 6. Ser igual de sensible para todos los picos eluidos o registrar únicamente aquellos picos que sean de interés. No ser afectado por cambios de temperatura o composición de la fase móvil. Ser capaz de monitorizar trazas de las especies a analizar. No contribuir de forma apreciable al ensanchamiento de los picos. Proporcionar una respuesta rápida que le permita detectar picos cromatográficos estrechos provenientes de la columna. Ser robusto y fácil de mantener. Terminología y Conceptos Generales Selectividad Un Detector no selectivo reacciona frente a una propiedad general del eluyente que pasa a través de él. Cuando un analito eluye de la columna, las características de esa propiedad cambian, y ese cambio bruto se mide y se registra. Esta detección no-selectiva es característica del detector de índice de refracción. En contraste, los detectores selectivos no reaccionan simplemente a los cambios en las propiedades generales en el eluyente sino que miden una respuesta definida que se produce por cambios en una propiedad específica o característica del analito. Dicha detección selectiva es característica de los detectores de Absorbancia UV, Fluorescencia y Espectrometría de Masas. Sensibilidad La señal más baja detectable no debe ser nunca menor de tres veces la altura del ruido promedio en la línea de base; Es decir, una relación señal a ruido (S/N) de 3:1 para un analito determinado determina su "Limite de Deteccion". Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 104 - Incidencias y Mantenimiento en HPLC Si la cantidad de analito inyectado cae por debajo de su límite de detección, entonces la señal no podrá diferenciarse del ruido. Para los análisis cuantitativos se recomienda una relación señal/ruido de 10:1 y define el “Límite de Cuantificación”. Figura mostrando una S/N de 10:1 − El Límite de Cuantificación (LoQ). A continuación se incluye un cuadro comparativo de la selectividad y sensibilidad ofrecida por diferentes detectores: Tipo de Detector Selectividad Sensibilidad (Min. Masa Detectada) Indice de Refracción Baja 1−5µg Conductividad Baja 10−50ng Light Scattering Evaporativo Baja 0.1−1.0ng Media 0.5−1.0ng Electroquímico Alta 50−500pg Fluorescencia Alta 10−100pg Espectrómetro de Masas Alta 1−25pg UV/Visible Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 105 - Incidencias y Mantenimiento en HPLC Rango Lineal El Detector ideal debería proporcionar una señal de respuesta cuya área fuera proporcional a la cantidad de muestra inyectada, independientemente de si está cantidad es grande o pequeña. Esta linealidad no es infinita, pero debe extenderse en el rango más amplio posible. La pendiente de la recta en el diagrama de abajo se denomina respuesta y representa en que magnitud el detector responde a un cambio específico en la concentración de la muestra: Figura mostrando la respuesta de un detector lineal y uno no lineal ¿Area o Altura de Pico? Es una práctica común emplear el área de pico cromatográfico para propósitos cuantitativos ya que proporciona una medida más robusta. Sin embargo, en ocasiones la altura de pico puede proporcionarnos resultados más exactos y reproducible, como es el caso de una pareja de picos crítica que o esté bien resuelta: Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 106 - Incidencias y Mantenimiento en HPLC 9.2 El Detector UV/Visible Principio Operacional − Detector UV/Visible Sintonizable Con el Detector UV/Vis Sintonizable, o de longitud de onda variable, la longitud de onda requerida se pre-selecciona por el analista por medio de software. Esta selección produce el movimiento físico de una red de difracción (monocromador) alojado en el interior del detector, permitiendo que un haz con la longitud de onda de la luz seleccionada ilumine el camino óptico de la celda de flujo y llegue al fotodiodo. El fotodiodo mide el cambio de absorbancia de los componentes de la muestra que pasan a través de la celda de flujo enviando una señal de respuesta al sistema de registro. Beam Splitter Detector UV/Visible de Longitud de Onda Variable (Sintonizable) Principio Operacional − Detector UV/Visible Diodo-Array A diferencia del detector sintonizable, el detector UV/Visible Diodo-Array (DAD) permite que pasen a través de la celda el rango completo de luz proveniente de la fuente. Esta luz se divide posteriormente en su espectro por medio de la red de difracción alcanzando la serie de fotodiodos, cada uno de ellos capaz de medir con precisión una porción extremadamente definida del espectro electromagnético de la luz incidente. Esta distribución óptica permite obtener mayor información que la obtenida simplemente con un análisis cromatográfico a una única longitud de onda. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 107 - Incidencias y Mantenimiento en HPLC Detector UV/Visible Diodo-Array (DAD) Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 108 - Incidencias y Mantenimiento en HPLC 9.3 Incidencias y Mantenimiento del Detector UV/Visible Lámparas de Deuterio y Tungsteno Una lámpara de Deuterio produce radiación intensa en el rango de longitudes de onda de 190−400nm gracias a una descarga de plasma realizada en una atmósfera de Deuterio (D2 ó “Hidrógeno Pesado”). Las Lámparas de Tungsteno se usan a menudo junto a lámparas de Deuterio, y emiten luz en el UV cercano y en la región visible del espectro electromagnético, típicamente en el rango de longitudes de onda de 340850 nm. La vida útil de cualquier lámpara de detector es necesariamente función del número total de horas en que una lámpara en particular puede realizar su trabajo de forma útil. En concreto, la vida útil de una lámpara será proporcionalmente más corta si la aplicación a que se dedica es a un análisis de trazas muy exigente más que a un método de ensayo, ya que una vez que el ruido de la línea de base o la deriva excedan los límites establecidos por el método, la lámpara deberá ser sustituida. Lineas de base con Ruido o Deriva indicativas de una lámpara gastada Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 109 - Incidencias y Mantenimiento en HPLC La emisión de una lámpara de Deuterio decrece de forma exponencial. En concreto, ésta emisión decrece lentamente hasta el final de la vida de la lámpara, momento en que comienza a decrecer rápidamente produciéndose finalmente un fallo en su encendido. Este rápido deterioro en la emisión puede proporcionar al usuario un aviso sobre el inminente fallo de la lámpara. A medida que la emisión de la lámpara se reduce se producirá una disminución en la relación S/N y en consecuencia no se obtendrá la sensibilidad a los analitos requerida por el método. Una lámpara de deuterio puede considerarse inutilizable cuando su emisión cae por debajo de del 50% de su valor original a una longitud de onda específica, o cuando el test de idoneidad del sistema (system suitability) no cumple el valor de S/N especificado. Existen tres razones fundamentales del decrecimiento de la emisión con el tiempo: • • • Evaporación del Tungsteno, Molibdeno o recubrimiento del filamento, que produce una emisión reducida o problemas en el encendido. Reacción del material de recubrimiento con la envoltura de cuarzo. Solarización de la envoltura de cuarzo a 200−250nm, provocando el desarrollo de absorción entre éstas longitudes de onda. La vida útil de una lámpara de deuterio es inversamente proporcional al número de encendidos que experimenta. Mantener la lámpara continuamente encendida aumentará su vida útil unas tres veces, en un promedio de 8 horas diarias de funcionamiento. Encender y apagar la lámpara en los descansos entre análisis o durante la noche reducirá apreciablemente su vida útil, siendo al final un procedimiento improductivo ya que la lámpara requerirá un tiempo de calentamiento de aproximadamente 30 minutos para estabilizar la línea de base. ¿Cuál es la vida media esperada de una lámpara de deuterio? Típicamente entre 800 y 1000 horas, ó 1800-2000 horas en las lámparas de deuterio de larga duración. La fuente de Tungsteno de la porción visible de muchos detectores UV/Visible tiene una vida extraordinariamente larga y en general falla porque se funde. Es importante resaltar que las lámparas de deuterio tienen caducidad en su almacenamiento. La lámpara puede envejecer considerablemente durante su almacenamiento, de forma que pueda ser inutilizable para su uso en un detector después de sólo 6 meses. Se recomienda por lo tanto pedir una lámpara de repuesto cuando estemos cerca del fin de su vida útil más que disponer permanentemente de una lámpara de repuesto a mano. Muchos softwares actuales disponen de una función de diagnóstico incorporada que nos permite medir la intensidad y el envejecimiento de la lámpara. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 110 - Incidencias y Mantenimiento en HPLC La Celda de Flujo La contaminación de la propia celda o de sus ventanas puede ser el resultado de una limpieza inadecuada del sistema o del tipo de muestra en sí misma. Unas ventanas de la celda que estén sucias producirán una pérdida progresiva tanto en la intensidad de la luz incidente como en la saliente, afectando así a la absorbancia de los analitos y a su respuesta en función del tiempo. Pérdida de sensibilidad causada por unas ventanas de la celda sucias Cualquier contribución a la absorbancia generada por la propia celda puede comprobarse y medirse por medio de un test específico de intensidad: típicamente esto implica el llenado de la celda con H2O o un solvente de referencia. Los siguientes procedimientos genéricos de limpieza de la celda de flujo se incluyen como suplemento a las recomendaciones específicas del fabricante para cada detector en particular; por favor, consulte los manuales de operación antes de emplear cualquiera de estos procedimientos. Los procedimientos generales de limpieza son aplicables a todos los detectores ópticos y pueden tener la forma de alguno de los siguientes: • • • Backflushing (limpieza en contracorriente) Limpieza con disolvente Límpieza con ácido La limpieza de la celda de flujo en contracorriente (Backflushing) elimina con frecuencia material particulado que pudiera haber bloqueado el tubo de entrada. Se conecta la bomba a la salida del detector y se invierte el flujo de eluyente a través de la celda, enviando el flujo directamente al contenedor de desechos. Si se observaban síntomas de una contrapresión alta, al invertir el flujo, la presión caerá y se eliminará el bloqueo. Este procedimiento debe aplicarse con precaución puesto que una presión excesiva sobre la celda de flujo podría provocar fugas alrededor de las Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 111 - Incidencias y Mantenimiento en HPLC ventanas o incluso la rotura de la propia celda de cuarzo. Para evitar la sobrepresión en la celda hemos de seleccionar la presión máxima de la bomba por debajo de la máxima presión que admita la celda e ir incrementando gradualmente el flujo en pasos de 0.1mL/min para evitar choques de presión. Una celda estándar de 1.0 cm de paso óptico suele tener una presión máxima de alrededor de 120 Bar. La limpieza con disolventes es útil cuando nos encontramos en presencia de un contaminante orgánico soluble o se sospecha que hay gotas de un solvente inmiscible en la celda de flujo. Pasando a través de la celda una serie de solventes con propiedades físicas y químicas diferentes p.ej. viscosidad, polaridad, etc, puede eliminarse de forma eficiente cualquier contaminante en un amplio rango de solubilidades. Dichas series de disolventes pueden consistir en Metanol, seguido de Tetrahidrofurano (THF), Cloruro de Metileno antes de volver de nuevo al Metanol. La limpieza ácida es el más agresivo de los tres procedimientos de limpieza descritos. Este procedimiento implica el paso de un ácido por la celda del detector para eliminar contaminantes y posterior paso de H2O para eliminar todos los restos de ácido. Para asegurar la eliminación de cualquier traza de residuo de ácido, y cualquier residuo potencialmente absorbente en el UV a bajas longitudes de onda, es necesario bombear H2O a través de la celda de flujo. Se utiliza preferentemente Acido Nítrico ya que los ácidos halogenados p.ej. HCl atacan agresivamente los componentes de acero inoxidable de la celda. Si ninguno de los métodos de limpieza anteriores eliminan la contaminación o los problemas de bloqueo, la celda tiene que desmontarse o reemplazarse. Existen Kits de Reconstrucción que son normalmente más económicos que adquirir una celda completamente nueva. Consulte el manual de operación para las recomendaciones y advertencias específicas. El paso de burbujas de aire a través de la celda causará picos de ruido (spikes) en el cromatograma. La desgasificación incorrecta del eluyente es por lo general la causa de los problemas con burbujas, especialmente si se ha utilizado mezcla a baja presión. La reducción de presión resultante de la mezcla de disolventes requiere una desgasificación adicional puesto que el aire es menos soluble en la mezcla de solventes que en el solvente puro original. Una celda de flujo sucia puede atrapar microburbujas cuando pasan a través de ella. Estas microburbujas pasan normalmente a través de la celda sin ser notadas pero cuando se atrapan pueden asociarse para generar burbujas mayores afectando al ruido y la deriva de línea de base. Cualquier burbuja atrapada debe purgarse de la celda requiriendo en ocasiones el uso de isopropanol, cuya mayor viscosidad comparada con el acetonitrilo o metanol, ayuda a arrastrar la burbuja enviándola a desechos. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 112 - Incidencias y Mantenimiento en HPLC Una lectura de absorbancia que se va fuera de escala de forma inmediata y evidente puede ser el resultado de una ventana de celda rota. El Solvente que pasa a través de la celda de flujo puede fugar hacia la superficie externa de la ventana provocando dichos cambios de absorción. La Red de Difracción (Monocromador) En la mayoría de los detectores UV/Visible de longitud de onda variable el monocromador viene pre-alineado de fábrica, y está encerrado en una unidad óptica sellada junto a algunos espejos que conforman el camino óptico. Un fallo en el monocromador se evidencia porque es incapaz de fijar la longitud de onda requerida para el análisis y requerirá de la intervención de un ingeniero de servicio técnico. Debe comprobarse la exactitud de longitud de onda bien empleando una solución de calibración conocida con un máximo de absorción definido, o bien, empleando una banda característica de emisión de la lámpara de deuterio. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 113 - Incidencias y Mantenimiento en HPLC 9.4 Picos Negativos La aparición de picos negativos se produce debido a factores no relacionados con las interacciones entre los analitos y la columna. Se observan picos Negativos cuando los cables de señal del detector están invertidos, o cuando el interruptor de polaridad se encuentra en la posición incorrecta. Los picos Negativos son normales cuando se utiliza el Detector de Indice de Refracción Los picos Negativos aparecerán en el cromatograma si la absorbancia de la muestra es menor que la de la fase móvil a la longitud de onda de detección. Hay que emplear por lo tanto fases móviles que muestren una absorbancia UV/Vis baja, o cambiar la longitud de onda de detección del analito. Si a lo largo del tiempo se concentran en la fase móvil compuestos fuertemente absorbentes puede aparecer una situación similar si el disolvente se recicla. Si está empleando reciclado de fase móvil y comienzan a aparecer picos negativos tras un uso prolongado de la fase móvil, lave abundantemente la columna con un disolvente fuerte y re-equilíbrela, con fase móvil recientemente preparada. Si desaparece el problema de picos negativos, es un indicio de que la fase móvil estaba contaminada. Cuado se emplea el detector de Diodo Array (DAD) es recomendable emplear una longitud de onda de referencia para posibilitar la sustracción de línea de base. Esto tiene la ventaja de corregir una línea de base con deriva mejorando así los límites de detección y de cuantificación. Es importante asegurarse de que la longitud de onda de referencia y el ancho de banda (bandwith) se han elegido correctamente para prevenir la posibilidad de que se produzca una absorbancia mayor que la de los analitos a detectar y se generen picos negativos en el cromatograma. El procedimiento correcto para la selección de las longitudes de onda de detección y de referencia se describe en relación con el espectro UV del ácido anísico. Para optimizar la detección de la menor cantidad posible de ácido anísico seleccione la longitud de onda de medida al pico más intenso Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 114 - Incidencias y Mantenimiento en HPLC del espectro, es decir, 252nm. Tratando el espectro como si fuera un pico pseudo-cromatográfico, compruebe su anchura a media altura (PWHH). Este será en ancho de banda de la longitud de onda de medida (Bandwith) p.ej. 30nm. Es posible reducir los efectos de fondo gracias a una adecuada elección de las longitudes de onda de medida y de referencia. La longitud de onda de referencia puede emplearse para eliminar los siguientes efectos, que provocarán todos ellos derivas de línea de base durante el análisis cromatográfico o bien mostrarán ondulaciones o ruido en la misma; 1. 2. Reducción del ruido generado por: • Particulas en suspensión • Cambios en el índice de refracción del eluyente cuando se emplean gradientes de elución. • Turbulencias en la celda de flujo Reducción en la deriva del instrumento causada por: • Envejecimiento / deformacion de la lámpara Identifique en el cromatograma el pico que absorbe a la longitud de onda mayor, en el ejemplo es el ácido anísico. Determine la longitud de onda a la que el pico del ácido anísico deja de absorber, se considera generalmente cuando la absorbancia cae por debajo de 1.0mAU, y en el ejemplo del ácido anísico será 300nm. Para evitar la posibilidad de una contribución a la absorbancia de la longitud de onda de referencia elegida, añada 10nm a este valor − Esta longitud de onda será el límite inferior de la longitud de onda de referencia, i.e. 310nm. Para eliminar los efectos de fondo descritos hay que elegir un ancho de banda grande, típicamente de 100nm, y situar la longitud de onda de referencia en el punto medio de esta ventana i.e. 360nm. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 115 - Incidencias y Mantenimiento en HPLC 9.5 El Detector de Light Scattering Evaporativo Principio de Operación − Detector Light Scattering Evaporativo El Detector de Light Scattering Evaporativo (ELSD) mide la cantidad absoluta de luz dispersada por las partículas de analito que se secan mediante evaporación. Siempre que la muestra esté presente en la cantidad suficiente, no se evapore por sí misma, o se descomponga durante la etapa de secado, el detector proporcionará una respuesta medible. El mecanismo de detección de Light Scattering Evaporativo implica tres etapas: 1. 2. 3. Nebulización, utilizando N2 u otro gas inerte, para formar un aerosol o pluma de gotitas uniformes. Evaporación del eluyente en un tubo de evaporación con temperatura controlada e independiente, generando una pluma de partículas de analito no-volátiles. Detección Óptica de la luz dispersada por las partículas de analito de un haz de luz incidente. La respuesta es proporcional a la masa de analito que pasa a través del haz de luz, y es independiente de cualquier propiedad espectral del analito. El Detector de Light Scattering Evaporativo El ELSD puede considerarse universal, y representa técnica de detección ideal para todos los compuestos no volátiles, un amplio rango de compuestos volátiles con la adecuada selección del eluyente, y aquellos compuestos que no poseen cromóforos UV/visible o varían mucho en sus coeficientes de extinción. Su sensibilidad es superior a la ofrecida por los detectores de índice de refracción, y se obtiene sin los problemas de deriva Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 116 - Incidencias y Mantenimiento en HPLC de línea de base, gracias a la independencia de la detección de las características espectrales de absorción del eluyente. La medida cuantitativa de la luz dispersada por una solución se realiza habitualmente con iluminación láser. La luz dispersada se mide como función del ángulo entre el detector y la dirección del haz incidente. En consecuencia, las medidas realizadas simultáneamente en un amplio rango de ángulos de dispersión permiten la determinación directa de la masa molar y el tamaño de las moléculas en solución, sin necesidad de generar una curva de calibración basada en referencia o suposiciones físicas sobre la muestra. Si es necesario, el analista puede crear una curva de calibración universal para un único analito, con la que puede interpolarse la concentración de todos los analitos de la misma clase. Incidencias y Mantenimiento del Detector de Light-Scattering Evaporativo La Celda de Detección Eventualmente la celda de detección de cada instrumento de light scattering se ensuciará: esto es especialmente cierto para la cromatografía acuosa. Detenga el flujo de fase móvil, y aumente el flujo de gas de nebulización para arrastrar cualquier partícula no volátil del interior de la celda. Por favor, consulte el manual de operación antes de realizar cualquier procedimiento de limpieza. Cualquier fase móvil que se utilice sin filtrar o tampones que empleen sales o modificadores no volátiles contribuirán a un ruido excesivo de línea de base o a picos continuos (spikes). En el peor de los escenarios dichos aditivos pueden contaminar el tubo de deriva del detector y la celda de detección. Modificadores volátiles que son aceptables incluyen: Ácido Trifluoroacético, Ácido Acético, Formiato Amónico, Acetato Amónico e Hidróxido Amónico. Si la señal del detector de light-scattering evaporativo está fuera de escala mientras la de la traza UV correspondiente es normal, esa respuesta aumentada es debida a compuestos que no absorben en el UV pero que eluyen de la columna. Recuerde que el ELSD responderá a todos los analitos semi-volátiles y no-volátiles. Ruido de Línea de Base Debido a una temperatura en el tubo de evaporación insuficiente, el ruido en la línea de base cromatográfica puede aumentar como resultado de una evaporación de fase móvil ineficaz. Aumente la temperatura de evaporación en pasos de 5ºC tanto como se necesite para localizar la temperatura más baja a la que se evaporará la fase móvil de forma eficiente, pero que minimice la pérdida de cualquier componente semi-volátil de la muestra. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 117 - Incidencias y Mantenimiento en HPLC Cuando se realizan gradientes de elución optimice la temperatura de evaporación para la condición de solvente que representa la mayor carga de evaporación. En análisis con Gradiente por Fase Reversa significará que la alta proporción de H2O al inicio del gradiente requerirá una temperatura de evaporación mayor que la correspondiente a una alta proporción de solvente orgánico. Este proceso se invertirá cuando se realicen análisis con gradiente en Fase Normal. Un nebulizador parcialmente bloqueado o dañado, y la degradación de la columna por exposición a disolventes incompatibles o condiciones extremas de pH pueden contribuir a elevados niveles de ruido. 9.6 El Detector Espectrométrico de Masas Los Espectrómetros de Masas trabajan ionizando las moléculas y analizando e identificando posteriormente estos iones según sus respectivos ratios masa/carga (m/z). Los dos componentes en este proceso son: 1. 2. La Fuente de Iones, donde se generan los iones. El analizador de Masa, que ordena los iones según su ratio m/z. Existen diferentes tipos de Fuentes de iones utilizadas comúnmente en Cromatografía Líquida/Espectrometría de Masas (LC/MS), siendo cada una de ellas adecuada para diferentes grupos de compuestos. Muchos de los recientes avances en LC/MS se han producido en el diseño de las Fuentes de iones y en las técnicas para ionizar las moléculas de analito que separan los iones resultantes de la fase móvil. La introducción de las técnicas de Ionizacion a Presión Atmosférica (API) ha permitido expandir el número y el tipo de los compuestos analizables que pueden ionizarse de forma efectiva. Durante el proceso API, las moléculas de analitos se ionizan en primer lugar, antes de ser mecánica y electrostáticamente “filtradas” de las especies neutras. Las técnicas comunes API son: 1. 2. 3. Ionización por Electrospray (ESI). Ionización Química a Presión Atmosférica (APcI). Fotoionización a Presión Atmosférica (APPI). Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 118 - Incidencias y Mantenimiento en HPLC Aplicabilidad relativa de los analitos a las técnicas API. Principio Operacional − Ionización por Electrospray (ESI) ESI utiliza la química en solución para pre-formar y mantener los iones de analito antes de que alcancen al espectrómetro de masas. El flujo de eluyente se convierte inicialmente en un aerosol (nebulización) en la fuente a presión atmosférica. Mientras el aerosol se encuentra en la fuente está sometido a un fuerte campo electrostático y a una corriente caliente de gas de secado inerte. El campo electrostático de alto potencial induce una posterior disociación de las moléculas de analito, mientras que el gas de secado reduce progresivamente las gotas de aerosol por evaporación del eluyente. Al reducirse el tamaño de las gotas, la concentración de la carga aumenta y llegado un cierto tamaño se alcanza el límite Rayleigh de inestabilidad generándose a su vez grupos de gotas cada vez más pequeñas. Eventualmente, el tamaño de gota se reduce tanto que las fuerzas de repulsión entre los iones de la misma carga exceden la tensión superficial de la gota, y son desorbidos directamente en fase gaseosa a través de una serie de explosiones de Coulomb. Estos iones son atraídos y conducidos a través de un capilar, unos conos de muestreo, y lentes skimmer hacia el analizador de masas para su posterior detección. Desde que los iones son desorbidos de las gotas hasta que alcanzan al analizador de masas, pueden aparecer una gran variedad de reacciones en fase gaseosa, típicamente transferencia de protones e intercambio de cargas. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 119 - Incidencias y Mantenimiento en HPLC Figura ilustrando el proceso de ionización por Electrospray Cualquier compuesto capaz de formar iones en solución, bien sea a través de un efecto de inducción de carga o bien debido a la existencia de algún grupo ionizable en su estructura, es susceptible de ser analizado por ESI. ESI es especialmente útil para el análisis de grandes biomoléculas, como las proteínas, oligonucleótidos y carbohidratos. Estas grandes moléculas adquieren múltiples cargas y, a través de un proceso denominado Deconvolución, esa serie de señales de múltiple carga puede convertirse y asignarse a una única carga en una escala de masa real. ESI puede también emplearse para analizar pequeñas moléculas farmaceúticas como benzodiacepinas u otras moléculas de interés biológico. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 120 - Incidencias y Mantenimiento en HPLC Principio de Operación − Ionización Química a Presión Atmosférica En APcI el eluyente se vaporiza en un nebulizador calentado, típicamente a 350-500ºC. El calor causa la vaporización del eluyente a la fase gaseosa ionizándose sus moléculas posteriormente gracias a electrones que provienen de la descarga continua de una aguja de corona. Los iones de solvente formados transfieren entonces su carga a las moléculas de analito que son consecuentemente atraídas hacia el capilar de entrada y conducidas a través del mismo, de los conos de muestreo y de las lentes de skimmer hacia el analizador de masas para su posterior detección. Figura ilustrando el proceso de Ionización Química a Presión Atmosférica APcI se aplica a una gran variedad de analitos polares y no polares. Sin embargo, rara vez se producen los fenómenos de carga múltiple que son tan característicos de ESI, y en consecuencia su aplicabilidad está típicamente dirigida a moléculas de menos de 1500 Da de peso molecular que no sean térmicamente lábiles. En comparación con ESI, la máxima sensibilidad en APcI se consigue generalmente cuando se emplea cromatografía en fase normal, ya que su mecanismo de ionización favorece a las moléculas neutras en solución, y los disolventes empleados en este modo cromatográfico no mantienen fácilmente iones en solución. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 121 - Incidencias y Mantenimiento en HPLC Principio de Operación − Fotoionización a Presión Atmosférica APPI opera de forma similar a APcI. El eluyente LC se nebuliza en primer lugar con un vaporizador calentado pasando a la fase gaseosa. Los fotones emitidos en la fuente por una lámpara de descarga se controlan cuidadosamente con respecto a sus energías de ionización, permitiendo la ionización de tantas moléculas de analito como sea posible mientras se minimiza la ionización de las moléculas del eluyente. Los iones de analito formados son atraídos hacia el capilar de entrada, y conducidos a través del mismo, de los conos de muestreo y de las lentes de skimmer hacia el analizador de masas para su posterior detección. APPI se aplica a la mayoría de analitos analizables por APcI, pero muestra una sensibilidad mejorada para los compuestos altamente no-polares cuando se realiza la cromatografía a bajos flujos (<100µL/min). Incidencias y Mantenimiento del Espectrómetro de Masas Iones Contaminantes La contaminación de iones en el espectro de masas produce una pérdida de sensibilidad en los analitos y aumenta la complejidad en la asignación de las masas. Estos iones contaminantes pueden provenir de diferentes orígenes, incluyendo: 1. 2. 3. La Fase Móvil La mayoría de los solventes de grado HPLC contendrán impurezas que no absorban en el UV, o estén presentes a niveles de concentración por debajo de los límites de detección de la mayoría de otros detectores LC, pero pueden ser la mayor fuente de contaminación de iones observada en el espectro de masas. Las botellas de vidrio de H2O grado HPLC son extraordinariamente proclives a contener iones sodio y potasio que generan iones aducto de una intensidad significativa. Modificadores de la Fase Móvil. Muchos de los modificadores típicos de las fases móviles pueden generar por sí mismos iones fuertes, o introducir iones contaminantes en la fase móvil. Siempre que sea posible deben utilizarse reactivos de la mayor pureza posible, y minimizar la concentración empleada. Tubos de Conexión. Evite los tubos de plástico para minimizar la introducción de iones Ftalato. El ión de Ftalato 609Da es a menudo el pico base de cualquier espectro de masas, y puede ser introducido inadvertidamente desde los tapones con septum. Minimice la contribución de los Ftalatos purgando la fuente con N2 gas a temperaturas más altas de las utilizadas por término medio para eliminar la contaminación. Utilice tubo PEEK incoloro para reducir el ruido de fondo. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 122 - Incidencias y Mantenimiento en HPLC 4. 5. Los tubos de acero inoxidable a menudo producen adsorción de los analitos y contaminación con álcali. El Gas de Secado La contaminación por el gas de secado es especialmente importante en APcI, pues es extremadamente sensible a los mecanismos de ionización en fase gaseosa. La principal impureza en el N2 proveniente de cilindros o de Tanques Dewar es la existencia de hidrocarburos. La principal impureza en los generadores de N2 es el O2 con trazas de Ar, puesto que los prefiltros de los generadores atrapan los hidrocarburos. Línea de Exhaust (Escape) En algunas ocasiones es posible que algunos contaminantes fluyan en hacia la fuente provenientes de la línea de exhaust. Cuando la salida de exhaust de la fuente y la de la bomba de vacío externa están combinadas en una, es posible que algo del aceite de la bomba migre hacia la cámara de spray recubriendo los componentes de la fuente y generando contaminación del analizador de masas. Para evitarlo se puede incorporar una trampa intermedia entre la salida de la bomba y la fuente, manteniendo una presión positiva en la cámara de spray en todo momento. Es recomendable disponer de unos espectros de masas de fondo y de blanco obtenidos bajo condiciones estándar como referencia. Esto ayudará en la potencial identificación de fuentes de contaminación en situaciones futuras. Mantenimiento de la Bomba Rotatoria Las Bombas Rotatorias son responsables de proporcionar el vacío inicial, y se emplean para ayudar a la bomba turbomolecular de alto vacío unida directamente al espectrómetro de masas. El vacío inicial está típicamente en el rango de 1.5 − 2.5 Torr. Adicionalmente, la bomba rotatoria captura y atrapa solvente residual, contaminantes y analitos introducidos en el espectrómetro de masas. El nivel de fluido de la bomba rotatoria debe comprobarse una vez a la semana. Esto se hace normalmente por inspección visual del nivel de fluido en la ventana situada en el frontal de la bomba. El nivel de fluido debe estar entre las líneas marcadas de nivel máximo y mínimo. Si está cercano a la línea de mínimo, hay que añadir fluido o alternativamente sustituirlo todo si nos encontramos próximos la fecha de mantenimiento. Cuando está limpio, el fluido de la bomba tiene un color Amarillo pálido, que a través del tiempo y uso se va oscureciendo gradualmente. Cada seis meses, o incluso antes dependiendo del uso, es necesario cambiar el fluido de la bomba. Para éste cambio es necesario apagar y ventear el espectrómetro de masas según las instrucciones del fabricante. Eleve la bomba del suelo y coloque un contenedor para recoger el fluido viejo que saldrá al retirar el tapón de drenaje. Desenrosque y quite el tapón de la parte superior de la bomba y posteriormente el tapón de drenaje de Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 123 - Incidencias y Mantenimiento en HPLC la parte inferior del cuerpo de la bomba permitiendo que el aceite drene. El fluido drenará más rápido y más completamente si la bomba está aún caliente. Desechar el fluido antiguo para su tratamiento como residuo tóxico. Reinstale el tapón de drenaje y llene la bomba con fluido nuevo hasta que su nivel esté cerca pero no por encima de la línea superior marcada en la ventana de visualización. Aproximadamente una vez a la semana será necesario hacer un “ballast” de la bomba. El “ballasting” se realiza abriendo la válvula de “ballast” de la bomba rotatoria, lo que permite a la atmósfera entrar a través del fluido de la bomba y eliminar contaminantes atrapados restaurando su compresibilidad. Adicionalmente, este proceso permite que cualquier aceite atrapado en el filtro drene de nuevo al reservorio principal. El “Ballasting” debe realizarse únicamente cuando el instrumento no está en uso, ya que el vacío en el analizador estará necesariamente comprometido durante el proceso. No deje la bomba con la válvula de Ballast abierta durante largos periodos de tiempo ya que esto puede potencialmente provocar daños; 30 a 60 min deben ser suficientes dependiendo del uso del instrumento, flujos y tipo de tampones empleados. Figura ilustrando la posición de la válvula de gas “ballast” Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 124 - Incidencias y Mantenimiento en HPLC Tutorial 3 Pregunta 1 Evaluar los problemas mostrados en el siguiente cromatograma y sugerir las posibles causas de éstos y cómo pueden corregirse: Observaciones: Posibles Causa(s) y Solución(es): Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 125 - Incidencias y Mantenimiento en HPLC Pregunta 2 Usando los conocimientos adquiridos durante el curso complete un programa de mantenimiento para su sistema de HPLC empleando la tabla de abajo como guía. Componente Frecuencia de Mantenimiento. Cada…….. Procedimiento de Mantenimiento 48 Horas Semana Mes + Mes La Fase Móvil Solvente Tamponado Filtros de Solvente La Bomba Check Valves Pistones y Sellos Frita Válvula de Purga Cebado El Autoinyector Sello del Rotor Aguja y Asiento de la Aguja La columna Guarda Columna Columna El Detector Lámpara Celda Flujo Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 126 - Incidencias y Mantenimiento en HPLC 10. Guia para incidencias La interpretación del cromatograma HPLC es esencial para el diagnóstico correcto y la corrección de los problemas en LC. Los problemas se pueden identificar a partir del cromatograma y pueden englobarse en una de las tres categorías siguientes: • • • Líneas de Base La apariencia o el perfil de la línea de base pueden ayudar en la identificación de muchos de los problemas asociados con los componentes del sistema. Forma de los Picos La forma del pico cromatográfico proporciona una valiosa información al caracterizar las prestaciones proporcionadas tanto por los componentes del sistema como por la columna. Tiempos de Retención Cualquier variación observada en los tiempos de retención de los analitos es crítica en el diagnóstico para eliminar problemas del sistema LC. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 127 - Incidencias y Mantenimiento en HPLC 10.1 Líneas de Base La pariencia de la línea de base cromatográfica puede describirse de múltiples formas distintas. Estas se describen a continuación, haciendo referencia a las posibles causas y sus acciones correctoras. 10.1.1 Líneas de Base Erráticas Mostradas en el cromatograma como variaciones aleatorias a corto plazo (positivas y negativas), no muestran un patrón lógico cuando las estudiamos en un intervalo mayor de tiempo. La siguiente tabla enumera una serie de razones por las cuales la línea de básica puede ser errática, junto a la solución apropiada sugerida. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 128 - HPLC Troubleshooting and Maintenance Líneas de Base Erráticas Componente Botellas de Disolvente Causa Potencial Desgasificación mezcla incorrecta de la Fase Móvil Solución Asegurese de desgasificar convenientemente. Asegúrese que la Fase Móvil se ha preparado correctamente de acuerdo al PNT, incluyendo la(s) necesaria(s) etapa(s) de filtración. Bomba HPLC Fallos en la Válvula Proporcional de Gradiente (mezclado) Llene las 4 botellas de disolvente con el mismo volumen del mismo y realice una mezcla 1:1:1:1 durante un tiempo suficiente. El consumo en cada botella de disolvente debe ser el mismo si las válvulas individuales de cada canal operan correctamente. Válvulas antirretorno de entrada y/o salida pegadas o parcialmente bloqueadas. Sustituya y compruebe Realice los tests de mantenimiento específico indicados por el fabricante para evaluar la integridad de los componentes individuales del cabezal de la bomba. Limpie o sustituya los que sean necesarios siguiendo el protocolo descrito (ver pg. 46 ss), o las instrucciones del fabricante. Fugas en los sellos de los pistones Sustituya y compruebe. Automuestreador Adsorción de muestra en los componentes de la válvula de inyección. Columna Ligero “sangrado” de muestra fuertemente retenida o matriz Use un disolvente para la muestra lo más fuerte posible sin que comprometa la forma de pico (ver pgs. 65 ss). Implemente un régimen de lavados de la válvula tras cada inyección. Use una guarda columna. Asegurese de que la columna se lava con un disolvente fuerte después de cada muestra o lote de muestras (ver pgs. 86 ss). Detector Tiempo de Reequilibración insuficiente Mínimo 10 volúmenes de columna. En volúmenes de columna. Polvo o suciedad en los tarjetas electrónicas Sustituya el detector y compruebe. Si es necesario solicite una visita al servicio técnico. Lámpara cerca del fin de su vida útil. Mantenga un diario del uso de la lámpara. Celda de Flujo contaminada con muestra y/o matriz Captura de Datos Conversor Analógico-Digital o tarjetas electrónicas defectuosas métodos con Par Iónico 20−50 Establezca un tiempo obligatorio de sustitución de la lámpara p.ej. 80% del total de su vida útil esperada. Desmonte y limpie o sustituya la celda de flujo según las directrices del fabricante. Sustituya el sistema de captura de datos y compruebe. Si es necesario solicite una visita al servicio técnico. Conexiones con mala toma de tierra. Compruebe y ajuste todos los cables de conexión visibles. Perdída de contacto en los cables Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 129 - HPLC Troubleshooting and Maintenance 10.1.2 Línea de Base Cíclica (Corta Frecuencia) Se observa como una oscilación cíclica positiva o negativa y a corto plazo en la traza cromatográfica. Línea de base cíclica a corta frecuencia y línea de base normal No confunda una burbuja de aire en la celda del detector con un problema con la lámpara. Las burbujas de aire pueden fluir hacia el detector y causar un pico muy estrecho, o una perturbación que se parece a un pico cromatográfico, aunque de una ancho de pico apreciablemente menor. En otros casos la burbuja de aire puede pararse en la celda de flujo y causar un desplazamiento dramático de la línea de base; generalmente fuera de escala. La desgasificación inapropiada de los disolventes es por lo general la causa más frecuente de problemas con burbujas, y en especial si se está empleando un sistema de mezcla a baja presión. En estos casos la reducción de presión producida en la mezcla de los disolventes permite la desgasificación de los mismos al ser menos solubles en la mezcla que en los disolventes por separado. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 130 - HPLC Troubleshooting and Maintenance Si no lo tiene instalado, considere usar un regulador de contrapresión después del detector para evitar la desgasificación asociada a la reducción de presión. Elija un regulador de contrapresión que tenga una especificación de presión MENOR que la presión máxima que permite la celda de flujo del detector. Como alternativa puede usar un tubo PEEK de 50 cm de longitud y 0.17mm de d.i. a la salida del detector y al que se le conecta la línea de desechos. Es posible identificar problemas de burbujas de aire parando el flujo de la fase móvil, si el problema es una burbuja, la línea de base permanecerá estable, bien dentro ó fuera de escala. En cambio, un problema electrónico en la lámpara permanecerá cuando se pare el flujo de fase móvil. La tabla siguiente enumera diferentes razones por las que la línea de base del cromatograma puede mostrar oscilaciones de corta frecuencia junto con su solución adecuada. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 131 - HPLC Troubleshooting and Maintenance Línea de Base Cíclica (Corta Frecuencia) Componente Botellas de Disolvente Causa Potencial Desgasificación o mezcla inapropiada de la fase móvil Solución Asegurese de desgasificar convenientemente. Asegúrese que la Fase Móvil se ha preparado correctamente de acuerdo al PNT, incluyendo la(s) necesaria(s) etapa(s) de filtración. Bomba HPLC Aire atrapado en el cabezal(es) de la bomba Rellene las botellas de disolvente y cebe las líneas lo que se requiera. Abra la válvula de purga, aumente el flujo (5mL/min) para desplazar el aire atrapado. Válvulas antirretorno de entrada y/o salida pegadas o parcialmente bloqueadas. Afloje la rosca de la válvula de salida para desplazar el aire atrapado. Sustituya y compruebe. Limpie o sustituya si es necesario utilizando el protocolo genérico descrito (ver página 46 ss), o siguiendo las instruccines del fabricante. Automuestreador Sustituya o repare el amortiguador de pulsos (damper) Solicite una visita del servicio técnico si se requiere. Fuga potencial en el cuerpo de la válvula de inyección que introduce aire. Realice el test de mantenimiento especificado por el fabricante para evaluar la integridad de los componentes del automuestreador. Loop o conexión de la aguja potencialmente defectuosos. Revise la válvula conmutable para buscar conexiones flojas. Desmonte y re-asiente la válvula conmutable o sustituya sus comentes si es necesario. Detector Burbujas de aire atrapadas en la celda de flujo. Purge el aire. NO exceda el límite máximo de operación-típicamente 120 bar para una celda de flujo de 1 cm y 13µL de volumen. Revise la documentación del fabricante para confirmar. Si no se encuentra integrado en el detector, conecte un restrictor de contra presión para evitar la desgasificación. Típicamente un tubo de PEEK de 50cm de largo y 0.17mm d.i. será suficiente. Fluctuaciones de la corriente eléctrica de alimentación Traducción Grupo Biomaster 2009 Compruebe otros instrumentos fluctuaciones si es necesario. e instale protección © Crawford Scientific 2007 - 132 - contra las HPLC Troubleshooting and Maintenance 10.1.3 Línea de Base Cíclica (Larga Frecuencia) Observada como una oscilación positiva o negativa de la traza cromatográfica de larga frecuencia. La tabla siguiente enumera diferentes razones por las que la línea de base del cromatograma puede mostrar oscilaciones de larga frecuencia junto con su solución adecuada. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 133 - HPLC Troubleshooting and Maintenance Línea de Base Cíclica (Larga Frecuencia) Componente Causa Potencial Solución Botellas de Disolvente Fluctuaciones de Temperatura que cambian la composición del disolvente o su viscosidad Compruebe que la botella de disolvente está cerrada. Bomba de HPLC Aire atrapado en el cabezal(es) de la Bomba Abra la válvula de purga, aumente el flujo (5mL/min) para desplazar el aire atrapado. Columna Fluctuaciones en la temperatura del termostado de columnas Compruebe la calibración de temperatura del horno de columnas. Ligero “Sangrado” de una muestra o de matriz fuertemente retenida Asegúrese de que la columna se lava con un disolvente fuerte al final de cada muestra o lote de muestras analizadas. Burbuja(s) de aire atrapada(s) en la celda del detector Purge el aire. NO exceda el límite máximo de operación-típicamente 120 bar para una celda de flujo de 1 cm y 13µL de volumen. Mantenga constante la temperatura de la fase móvil. Afloje la rosca de la válvula de salida para desplazar el aire atrapado. Detector Revise la documentación del fabricante para confirmar. Si no se encuentra integrado en el detector, conecte un restrictor de contra presión para evitar la desgasificación. Típicamente un tubo de PEEK de 50cm de largo y 0.17mm d.i. será suficiente. Traducción Grupo Biomaster 2009 © Crawford Scientific - 134 - HPLC Troubleshooting and Maintenance La figura de abajo muestra como la termostatización de la fase móvil no solo asegura una línea de base aceptable sino que además mantiene una forma de adecuada del pico cromatográfico. El cromatograma (b) se obtuvo con una temperatura de columna de 380C, pero con la fase móvil entrando a temperatura ambiente (220C). Puede observarse una apreciable distorsión y ensanchamiento de los picos. El cromatograma (a) se obtuvo empleando un pre-calentador del eluyente, que consiste en un tubo de acero inoxidable de 0.25mm d.i. enrollado como una bobina plana y fijado al bloque de calentamiento del horno de columnas, situado entre el automuestreador y la columna. Algunos hornos comerciales incluyen ya un pre-calentador de fase móvil. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 135 - Incidencias y Mantenimiento en HPLC 10.1.4 Lineas de Bases con Deriva Se define como Deriva a la variación de la señal cromatográfica con el tiempo siempre en la misma dirección (tendencia), sea ésta positiva o negativa. La tabla siguiente indica diferentes razones por las que la línea de base del cromatograma puede mostrar deriva junto con su solución adecuada. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 136 - HPLC Troubleshooting and Maintenance Líneas de Base con Deriva Componente Botellas de Disolvente Causa Potencial Fuerte absorbacia mostrada por un(os) componete(s) de la fase móvil. Contaminación y/o Evaporación de la Fase Móvil. Solución Si se usa un Detector Diodo-Array (DAD) seleccione una longitud de onda de referencia y un ancho de banda adecuados para eliminar el efecto de absorbancia del (los) componente(s). Registre los nº de lote de los disolventes y aditivos para identificar fuentes potenciales de contaminantes que absorban. Prepare fase móvil fresca y compruebe de nuevo. Compruebe los puntos de ebullición de los componentes de la fase móvil. Tape las botellas de disolvente para evitar pérdidas por evaporación. Bomba HPLC Automuestreador Inmiscibilidad de los disolventes empleados en la Fase Móvil. Lave el sistema con un disolvente intermedio compatible antes de introducir nueva fase móvil. Evalúe la posible inmiscibilidad refiriéndose a la tabla de miscibilidad (Ver pág. 15). Columna no acondicionada con la nueva fase móvil. Cebe las líneas y lave el sistema de HPLC hasta que se obtenga una línea de base estable. Re-equilbrado insuficiente de la columna o del gradiente de elución. Si empleamos un gradiente completo (i.e. 5 − 100% solvente B), debemos asegurar un cambio de 10 volúmenes de columna antes de realizar una nueva inyección de muestra. Adsorción de muestra en los componentes de la válvula de inyección. Mantenga la posición de inyección de la válvula del inyector al menos unos minutos después de la introducción de la muestra para asegurar un lavado intenso. Use un disolvente de la muestra tan fuerte como sea posible sin que se comprometa la forma de pico Implemente un régimen de lavado de la válvula después de cada inyección. Columna Detector Ligero “sangrado” de muestra o matriz fuertemente retenida. Asegúrese de que la columna se lava con un disolvente fuerte después de cada muestra o lote de muestras. Método de Fase Reversa con formación de Pares Iónicos. Aumente el tiempo de re-equilibrado a 20-25 volúmenes. Exposición del Sistema gradientes de temperatura. Termostatice la columna y las líneas de entrada y salida de la misma. Dirección Negativa − Componente fuertemente absorbente en un eluyente débil. Seleccione en el DAD la longitud de onda de referencia y un ancho de banda adecuados para eliminar el efecto de absorbancia del (los) componente(s) Dirección Positiva − Componente fuertemente absorbente en un eluyente fuerte. Traducción Grupo Biomaster 2009 Compruebe que está usando el disolvente correcto. Asegúrese de que la polaridad de la señal del detector está correctamente seleccionada. © Crawford Scientific 2007 - 137 - HPLC Troubleshooting and Maintenance 10.1.5 Líneas de Base con Ruido Las perturbaciones momentáneas positivas o negativas o los saltos de la señal cromatográfica causando picos transitorios se denominan “spikes”. La forma más simple de comprobar si hay un excesivo ruido en la línea de base es realizar un cromatograma sin inyectar muestra. Amplíe el cromatograma blanco resultante de forma que pueda medir fácilmente la amplitud de la línea de base. Seleccione una región de ésta, p.ej. aproximadamente de un minuto, expandiéndola lo suficiente para mostrar el ruido, y trace líneas que describan a “grosso modo” los límites de éste. La distancia física entre ambas líneas puede convertirse a unidades de Absorbancia (AU). La mayoría de los detectores tienen especificaciones de ruido de 0.5 − 1.0 x 10-5 AU, de forma que un detector que muestre excesivo ruido puede identificarse con facilidad. Un ruido excesivo en la línea de base puede reducirse de forma aceptable gracias a la instalación de un filtro de © Crawford Scientific 2007 resitencia capacitancia. Las especificaciones del detector se suelen dar sobre una celda seca y vacía a 255nm. No es muy práctico tener que crear una celda seca, especialmente si se desea medir el ruido bajo condiciones similares a las del análisis cromatográfico. Es aceptable sustituir la técnica de la celda de flujo seca por una fase móvil isocrática y estacionaria. Si el ruido observado en estas condiciones está aproximadamente a 2x de la especificación, el detector está funcionado correctamente. El ruido a λs más bajas, como 215nm, será mayor aquel mostrado a 255nm con la fase móvil fluyendo a través de la celda. Los transitorios (“Spikes”) se caracterizan por tener un ancho de pico a mitad de altura considerablemente menor que un pico cromatográfico real. Aparecerán de forma aleatoria durante el análisis cromatográfico, estando por encima del ruido de línea de base determinado de la forma anteriormente indicada. Una lámpara de detector cerca del límite de su vida o un ruido electrónico excesivo generalmente causan este tipo de perturbaciones. La tabla siguiente sugiere diferentes causas para que una línea de base muestre ruido junto con su solución propuesta. - 138 - HPLC Troubleshooting and Maintenance Líneas de Base Ruidosas Componente Causa Potencial Solución Botellas de Disolvente Burbujas de aire en la Fase móvil Aségurese de desgasificar adecuadamente Bomba HPLC Compruebe todas la conexiones buscando fugas que pudieran provocar la entrada de aire Elimine está posibilidad parando el flujo de fase móvil: si aún se muestra el ruido entonces el conjunto de suministro de disolvente no es la causa. Compruebe las áreas alrededor del cabezal de la bomba por si hubiera depósitos de sales de los támpones. Apriete las conexiones si es necesario. Automuestreador Introducción de aire durante la inyección. Realice los test de mantenimiento especificados por el fabricante para evaluar la integridad de los componentes individuales del Automuestreador. Disolvente de la muestra o Fase móvil incorrectas Columna Sustituya los componentes dudosos si es necesario. © Crawford Scientific 2007 Si la viscosidad del disolvente de la muestra es alta, o posee un bajo punto de ebullición, es conveniente reducir la velocidad de succión de muestra y la de inyección para evitar cavitación. Temperatura de la Columna mayor que el punto de ebullición de alguno de los componentes de la fase móvil. Pérdida de los ligandos de la fase estacionaria. Disminuya la temperatura de separación para evitar la ebullición del disolvente. Crawford Scientific Revalide el método si hay una pérdida de©resolución o cambia la selectividad de la columna. Compruebe que el método está operando en el rango correcto de pH para esa columna. Compruebe que el método funciona dentro de los límites de especificados para esa columna. Disolución de la sílice de la fase estacionaria. Detector Pertubaciones electrónicos. eléctricas “spikes” provenientes de los circuitos pH Sustituya el detector y compruebe. Solicite una visita del servicio técnico si se requiere. Burbujas de aire atrapadas en la celda del detector. Purge el aire. NO EXCEDA el límite superior de presión establecido para la celda del detector, típicamente de 120 bares para una celda estándar de 1cm de paso óptico y 13µL de volúmen. Revise la literatura del fabricante para confirmarlo. Si no se encuentra integrado en el detector, conecte un restrictor de contra presión para evitar la desgasificación. Típicamente un tubo de PEEK de 50cm de largo y 0.17mm d.i. será suficiente. Mantenga un registro de las horas de uso de la lámpara. Captura de Datos Lámpara cerca del límite de su vida útil. Establezca un periodo de sustitución obligatorio de la lámpara. P.ej. al 80% de su tiempo de vida útil esperada. Mala toma de tierra / Conexión floja Sustituya el sistema de adquisición de datos y compruebe. Revise y reconecte todos los cables de conexión visibles. - 139 - 2007 HPLC Troubleshooting and Maintenance 10.2 Formas de Pico 10.2.1 Ensanchamiento de los Picos La corrección de picos ensanchados requiere determinar si dicho ensanchamiento es producto de cambios en el sistema HPLC, deterioro de la columna o se trata de un “eluido tardío” proveniente de una inyección anterior. Si todos los picos separados se han ensanchado, siendo los primeros los que muestren más ensanchamiento, el problema vendrá causado simplemente por la introducción de un gran volumen extra-columna en el sistema; • • • Adición de una longitud de tubo desproporcionada. Una conexión de capilar o de columna deficiente. Conexiones de PEEK (ajustables a mano) desgastadas. Si no es así, o el ensanchamiento de pico se viene observando durante un periodo de tiempo largo, es un indicativo del deterioro general de la propia columna. La Eficiencia de la Columna, o número de platos (N), se usa como medida matemática del deterioro de una columna en función del tiempo y del número de inyecciones. Midiendo el número de platos para un determinado compuesto de la muestra, o evaluando el pico cromatográfico con un conjunto de estándares de calidad recomendado por el fabricante de la columna, y comparando el resultado con el registro histórico nos indicará la magnitud del deterioro. Siempre que la fase móvil y el resto de parámetros del sistema no se hayan cambiado, la retención de los analitos no debería variar significativamente cuando la eficiencia caiga. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 140 - HPLC Troubleshooting and Maintenance La Eficiencia de la columna se puede calcular aplicando la ecuación siguiente: N = 5.54 x (tR / w0.5)2 Dónde; N= Número de Platos de la Columna tR = Tiempo de Retención del Analito w0.5 = Ancho de pico a mitad de altura (FWHH) • • Si N aumenta cuando aumenta el tiempo de retención del analito, es que la columna genera la mayor contribución al volumen muerto del sistema, y el HPLC funciona correctamente. Si N decrece o llega a ser aleatorio cuando aumenta el tiempo de retención del analito, es el HPLC quien genera la mayor contribución al volumen muerto del sistema y no funciona adecuadamente. Hay que evaluar si otros componentes del sistema pueden también contribuir al ensanchamiento de los picos. Éstos incluyen guarda columnas deterioradas, o la introducción inadvertida de gran volumen extra-columna. Los compuestos de alto peso molecular, en especial las proteínas, pueden ensanchar sus picos en columnas de tamaños de poro < 80A; asegurese que usa un tamaño de poro > 300A cuando analice dichas especies químicas. Puede sospecharse de que un pico que eluye tarde provenga de una inyección previa cuando aparece como un único pico ancho en un cromatograma con el resto de los picos estrechos. Es importante destacar que este pico puede no aparecer en todas las inyecciones, y por lo tanto puede no ser observado inicialmente, especialmente en separaciones multicomponente complejas. Puesto que este “eluído tardío” en cuestión sufre simplemente de un aumento en su factor de capacidad (k’), una aproximación simple para identificar su origen es permitir que el cromatograma tenga una duración de muchas veces el tiempo de análisis normal. Una forma más eficiente para localizar a un pico de elución tardía es calcular primero su tiempo de retención real. Esta aproximación tiene la ventaja de que permite saber con que inyección en particular está realmente asociado el pico. En primer lugar determine el nº de platos de un pico determinado del cromatograma. Como ejemplo tomaremos un pico con tiempo de retención de 12min, y un ancho de pico a mitad de altura (PWHH) de 0.15min. A partir de la ecuación descrita anteriormente obtendríamos: N = 5.54 x (12 / 0.15)2 y N = 35,456 Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 © Crawford Scientific 2007 - 141 - HPLC Troubleshooting and Maintenance Resolviendo la ecuación para tR tendríamos; tR = 0.425 x (w0.5) x √N y tR = 12.0 Midiendo el ancho de pico a mitad de altura del último pico es posible predecir su tR. En este ejemplo supondremos que el ancho de pico problema es de 0.5min. Utilizando este ancho de pico, y el número de platos previamente determinado a partir del pico cromatográfico de 35.456, el tiempo de retención calculado es de 40min. Esto significa que el último pico que eluye está relacionado con una inyección realizada 40 min antes. Re-inyectando la muestra sospechosa y alterando el tiempo de análisis en consecuencia podemos confirmar este tiempo de retención. A menudo no es posible eliminar estos picos de elución tardía. Por lo tanto, en lugar de modificar las condiciones de separación, o esperar hasta que el pico eluya antes de hacer la siguiente inyección, es generalmente más conveniente modificar el tiempo de análisis de forma que el pico de elución tardío caiga en una zona irrelevante del cromatograma posterior. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 142 - HPLC Troubleshooting and Maintenance 10.2.2 Desdoblamiento de Picos y Hombros Si todos los picos del cromatograma son dobletes probablemente la columna ha desarrollado un vacío o canalizaciones en su inicio. La sustitución de la columna confirmará rápidamente el problema. Si se sospecha de la existencia de un vacío hay que desmontar la conexión de cabeza de columna y comprobar visualmente la misma. Si existe un vacío hay que rellenarlo con fase estacionaria, y si no, limpiar o sustituir la frita y probar la columna de nuevo para establecer si el problema fue originalmente causado por una frita parcialmente obstruida. Adicionalmente, invertir la columna y lavarla con un 100% de disolvente fuerte puede eliminar cualquier contaminación que bloquee la entrada de la columna enviándola directamente a desechos. Compruebe las instrucciones del fabricante para saber si la columna puede mantenerse en orientación de flujo inversa. Puesto que el bloqueo parcial de la frita de entrada de la columna está generado por depósitos de partículas existentes en la fase móvil, disolvente de la muestra o rotor seal, asegurese de filtrar adecuadamente e insertar un filtro de línea entre el inyector y la cabeza de columna. Si solamente uno de los picos del cromatograma se muestra como un doblete, podemos sospechar de la existencia de otro pico o interferencia que coeluye. Confirme la presencia de esta interferencia cambiando la longitud de onda del detector, o mejorando la resolución de la separación usando una columna que proporcione una mayor eficiencia, p.ej. una columna más larga o empacada con partículas de menor tamaño. Ajuste la selectividad mostrada por el sistema de separación frente a los compuestos que coeluyen alterando la composición o el tipo de la fase móvil, la temperatura de separación o la fase estacionaria de la columna. Use algún procedimiento previo de limpieza de la muestra p.ej. SPE para eliminar cualquier interferencia contaminante. Evalúe si un determinado pico que coleuye proviene de una inyección previa o no, inyectando un blanco y evaluando según el procedimiento descrito en la pág 141. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 © Crawford Scientific 2007 - 143 - HPLC Troubleshooting and Maintenance Lave de forma rutinaria la columna con un disolvente fuerte de alto poder de elución cuando realice separaciones isocráticas. Este efecto se muestra en la figura de abajo, donde la forma de pico de un grupo de drogas básicas son separadas isocráticamente en una fase móvil de 25mM Na2HPO4 (pH3.0):MeOH (60:40) y su forma se mejora significativamente tras un lavado de columna con 100% de acetonitrilo. Si se ha inyectado un volumen de muestra demasiado grande, y el solvente de la muestra y la fase móvil no han sido adecuadamente emparejados, se producirán dos equilibrios de distribución, generando un particionamiento diferencial de la muestra en la columna. La consecuencia de esto será la formación de dobletes o picos con hombros. Refererirnos a las reglas empíricas descritas en las págs. 79 ss en relación con el emparejado del volumen de inyección y la fuerza del disolvente de la muestra ayudará a eliminar este fenómeno. La siguiente figura ilustra el deterioro en la forma de pico debido a un inadecuado emparejamiento entre el disolvente de la muestra y la fase móvil, mostrando la inyección de una mezcla de cuatro componentes disuelta en diferentes disolventes y separada en una columna de fase reversa C18 bajo condiciones isocráticas de MeOH:H2O 60:40 (v/v). La mezcla test se separó en cada cromatograma disuelta en; • (a) − Fase Móvil • (b) − Tetrahidrofurano • (c) − Etanol • (d) − Butanol Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 144 - HPLC Troubleshooting and Maintenance El aumento de la fuerza de elución del disolvente produce una degradación apreciable de la forma de pico cromatográfico. Evalúe si la columna ha sido saturada en masa. Es posible determinar empíricamente la masa máxima que puede inyectarse en un determinado análisis empleando la ecuación siguiente: ωmax ≈ 50 x (1 − k' / k')2 x dc2 Dónde; ωmax = k' = dc = Masa de muestra en µg Factor de Capacidad Diámetro interno de la Columna en cm Esta ecuación no funciona bien para tamaños de partícula < 5µm, o si compuestos básicos muestran adsorción diferencial con la fase estacionaria. ωmax se refiere a cada compuesto independiente en la muestra y no a la masa total de muestra. Por lo tanto, si no hay componentes de la muestra por encima del 10% de la masa total de muestra ωmax será 10x el valor efectivo calculado. Si se requiere use una fase estacionaria de mayor capacidad, es decir, con un mayor % de carbono u mayor tamaño de poro, una columna con mayor diámetro o reduzca la cantidad total de muestra inyectada. Desmonte la válvula de inyección y revise el rotor seal en búsqueda de un posible desgaste, incluyendo roturas entrecuzadas, que pueden generar la presencia de una ruta de flujo inesperada en el inyector. Sustituya el rotor seal si es necesario, fijando un número máximo de inyecciones antes de sustituir periódicamente el mismo. Para más información refiérase a la sección 5.3 Incidencias del Automuestreador − "Efecto Memoria". Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 © Crawford Scientific 2007 - 145 - HPLC Troubleshooting and Maintenance 10.2.3 Picos con Cola La aparición de colas en los picos es una de las deformaciones cromatográficas más frecuentes. Se dice que un pico tiene cola cuando muestra una asimetría mayor de 1.2, aunque en ocasiones picos con valores de As tan altos como 1.5 puedan ser aceptables para una gran variedad de ensayos. El factor de asimetría de pico (π) se calcula según la siguiente ecuación: π = wb2 / wb1 Donde; wb2 = Ancho del pico en la línea de base después del centro del pico wb1 = Ancho del pico en la línea de base antes del centro del pico La principal causa de aparición de cola en un pico es la coincidencia de más de un mecanismo de retención. En las separaciones por Fase Reversa, el principal modo de retención de los analitos es a través de interacciones hidrofóbicas no-específicas con la fase estacionaria. Sin embargo, también son frecuentes las interacciones polares con grupos de silanoles residuales existentes en el soporte de sílice. Compuestos que posean en su estructura grupos amino o cualquier otro grupo básico, pueden interactuar fuertemente con los grupos silanoles ionizados, produciendo picos con cola. Este efecto se muestra en la figura siguiente a un pH >3.0. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 146 - HPLC Troubleshooting and Maintenance Estas interacciones secundarias pueden minimizarse si se realiza la separación a un pH más bajo, asegurando así la protonación completa de los citados grupos silanol residuales; Otra alternativa es la utilización de columnas “end-capped”. “End-capping” es el nombre que se da al proceso químico por el cual los grupos silanoles residuales de la fase estacionaria se transforman en otros grupos funcionales sustancialmente menos polares en su superficie. Esta modificación tiene el efecto de reducir las interacciones secundarias que puedan producirse potencialmente con moléculas de analitos polares. Los reactivos químicos empleados para el proceso de “end-capping” incluyen generalmente; • • Trimetilclorosilosano (TMCS) Hexametildisilazano (HMDS) Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 © Crawford Scientific 2007 - 147 - HPLC Troubleshooting and Maintenance El proceso de “End-capping” reduce las colas observadas con analitos polares, bloqueando de forma eficiente sus interacciones con los grupos silanol residuales que pudieran ser potencialmente ionizables. A pesar de ello, el “end-capping” sólo disminuye aproximadamente a un 50% el número de grupos silanol no reaccionados. Debido a factores de impedimento estérico este tipo de reacciones rara vez son eficientes en términos absolutos y en consecuencia una columna completamente “end capped” no es nunca tanto como debería! En el caso de que todos los picos cromatográficos muestren cola debe considerarse la posibilidad de que la columna se haya sobrecargado por un exceso de muestra. Evalúe aplicando la ecuación descrita previamente en la página 104. Si fuera necesario utilice una fase estacionaria de mayor capacidad p.ej. de mayor % carbono o tamaño de poro, utilice una columna de mayor diámetro, o reduzca la cantidad absoluta de muestra o volumen inyectado. Examine la columna por si se hubieran formado volúmenes muertos o se hubiera bloqueado parcialmente la frita de entrada. Sustituir la columna por otra nos confirmará rápidamente el problema. Si se sospecha que existe algún volumen muerto, quite las conexiones de cierre y revise la cabeza de columna. Si se observa algún hueco en el relleno cúbralo con fase estacionaria, o si no, limpie o sustituya la frita de entrada y pruebe la columna de nuevo para ver si el problema estaba originado o no por una frita parcialmente obstruida. En ocasiones, invertir la columna y Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 148 - HPLC Troubleshooting and Maintenance lavarla con un 100% de un disolvente fuerte también permite eliminar bloqueos y contaminación del filtro de entrada. Compruebe las instrucciones del fabricante para ver si la columna puede mantenerse en la orientación inversa de flujo o no. Si sólo uno, o algunos, de los picos cromatográficos muestran cola, considere la posibilidad de que la columna esté mostrando efectos de partición o retención secundarios. Reduzca la probabilidad de que cualquier analito potencialmente básico interactué con los grupos silanol residuales usando columnas fabricadas con sílice desactivada (Tipo B), que han sido completamente bloqueadas (“endcapped”). Como parte de cualquier procedimiento de desarrollo de métodos reduzca el pH de la fase móvil para suprimir la ionización de cualquier grupo silanol. En general, una fase móvil con pH <3 será suficiente para este propósito. Adicionalmente, el empleo de fases móviles a bajo pH asegura la protonación de los analitos ácidos, aumentando su retención y minimizando cualquier tipo de interacción iónica. La figura de abajo ilustra el efecto producido en las colas de pico al reducir el pH de separación. La mezcla de cinco fármacos básicos consiste en Fenilpropanolamina, Efedrina, Anfetamina, Metanfetamina y Fenteramina (Picos 1−5) respectivamente. El primer cromatograma se adquirió a un pH de la fase móvil de 7.0, y en estas condiciones de separación, el Pico 4, Metanfetamina muestra un factor de cola USP al 5% (USP Tf 5%) de 2.35. La disminución del pH de la fase móvil reduce la interacción de los analitos básicos con los grupos silanol residuales eliminado de esta forma la cola excesiva de los picos. El segundo cromatograma se adquirió con una fase móvil a pH 3.0 y el USP Tf (5%) de la Metanfetamina en estas condiciones se redujo a 1.33. La adición de Trietilamina (TEA) a la fase móvil en niveles de concentración de 5−10mM, también reduce o posiblemente elimina, la cola en compuestos básicos. La TEA compite con los analitos polares básicos por enlazarse con los grupos silanol residuales, produciendo una función de “pseudo endTraducción Grupo Biomaster 2009 © Crawford Scientific 2007 © Crawford Scientific 2007 - 149 - HPLC Troubleshooting and Maintenance capping”. Esta alternativa sin embargo es menos conveniente, ya que la fase estacionaria se altera irreversiblemente por la adición de tal modificador, afectando a la selectividad mostrada por el sistema de separación posteriormente. La cromatografía de pares de iones puede resolver el problema de las colas para analitos ácidos o básicos. Otra opción para la separación de especies iónicas podría ser emplear cromatografía de intercambio iónico. Desde un punto de visto práctico, generalmente sólo es posible utilizar el pH para suprimir la ionización ácida ya que la ionización básica requiere valor de pH típicamente mayores de 8, y a ese pH se puede producir la disolución de la sílice. Sin embargo, los fabricantes de columnas ofrecen versiones que permiten trabajar a un pH mayor, protegiendo la sílice de su disolución por medio de ligandos bi- y tri- dentados. Estos ligandos son en realidad dos o tres ligandos tradicionales que se han puenteado por medio de procesos químicos patentados antes de aplicarlos a la fase estacionaria. Su estructura puenteada permite proteger su superficie frente a valores extremos de pH. La Figura muestra ligandos bidentados y la protección ofrecida a la superficie de la sílice gracias a su estructura de puente. Un único pico con cola también podría producirse por un pequeño pico eluyendo inmediatamente después del pico cromatográfico de interés. La posibilidad de que esto ocurra es la razón primordial por la que las colas de pico nunca deben ignorarse. Como se describía en el apartado “desdoblamiento de picos y hombros”, podemos confirmar la presencia de un interferente cambiando la longitud de onda de detección, o mejorar la resolución de la separación empleando una columna de mayor eficiencia p.ej. una columna más larga o empaquetada con partículas de menor tamaño. Ajuste la selectividad del sistema frente a compuestos co-eluyentes variando la composición o el tipo de la fase móvil, la temperatura, o la fase estacionaria de la columna. Emplee algún procedimiento de “clean-up” p.ej. SPE para eliminar cualquier contaminante interferente. Evalúe si alguno de los picos coeluyentes pueden provenir de inyecciones anteriores haciendo una inyección en blanco y siguiendo el procedimiento descrito en “Ensanchamiento de Picos” de la pág. 105. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 150 - HPLC Troubleshooting and Maintenance 10.2.4 Picos con Frente Si todos los picos cromatográficos muestran frente debe considerarse la posibilidad de una sobrecarga de la columna por exceso de muestra inyectada. Evalúelo aplicando la ecuación descrita previamente en la pág. 104. Si fuera necesario utilice una fase estacionaria de mayor capacidad p.ej. de mayor % carbono o tamaño de poro, utilice una columna de mayor diámetro, o reduzca la cantidad absoluta de muestra o volumen inyectado. Si se ha inyectado un volumen de muestra excesivamente grande, y el solvente de la muestra y la fase móvil no están adecuadamente emparejados, pueden producirse dos equilibrios de distribución, provocando una partición diferencial en la columna. Refiérase a las reglas empíricas descritas en las páginas 54-56 en relación al adecuado emparejamiento de entre el volumen de inyección con la fuerza eluotrópica del disolvente de la muestra para eliminar éste fenómeno. Un único pico que muestre frente también puede deberse a un pequeño pico que eluya justo antes del pico cromatográfico de interés. Esta posibilidad es la principal razón por la que un pico con frente nunca debe ser ignorado. Como se describía en el apartado “desdoblamiento de picos y hombros”, podemos confirmar la presencia de un interferente cambiando la longitud de onda de detección, o mejorar la resolución de la separación empleando una columna de mayor eficiencia p.ej. una columna más larga o empaquetada con partículas de menor tamaño. Ajuste la selectividad del sistema frente a compuestos co-eluyentes variando la composición o el tipo de la fase móvil, la temperatura, o la fase estacionaria de la columna. Emplee algún procedimiento de “clean-up” p.ej. SPE para eliminar cualquier contaminante interferente. Evalúe si alguno de los picos coeluyentes pueden provenir de inyecciones anteriores haciendo una inyección en blanco y siguiendo el procedimiento descrito en la pág. 89. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 © Crawford Scientific 2007 - 151 - HPLC Troubleshooting and Maintenance 10.2.5 Efecto Memoria y Picos Fantasma Los picos fantasmas son picos que aparecen en el cromatograma, incluso cuando no se ha realizado ninguna inyección. Hay dos causas frecuentes para la aparición de picos fantasma: 1. Puede ser un pico de elución tardío proveniente de una inyección anterior. Debido a que tal pico habrá estado en la columna durante un tiempo apreciable, se mostrará como mucho más ancho que el resto de picos cromatográficos de su alrededor. La fuente potencial puede trazarse empleando el método de cálculo de tiempos de retención descrito anteriormente en la pg 141. Modifique el método según se necesite, incluyendo el lavado de la columna con un disolvente fuerte para eliminar cualquier contaminante potencial que estuviera fuertemente adsorbido. 2. Los picos fantasmas pueden derivarse de una fase móvil contaminada. Bajo condiciones isocráticas los picos pueden ser distintos, o se puede observar un desplazamiento de la línea de base sólo después de que el HPLC esté funcionando durante algún tiempo. En condiciones de gradiente los picos normalmente aparecen al mismo tiempo de retención, tanto en la inyección de un blanco como en la de una de muestra. En cualquier caso, dichos picos fantasma pueden trazarse como originados por una fase móvil contaminada. Para aislar la fuente contaminación use fase móvil recién preparada, sólo disolventes de grado HPLC, Agua de grado HPLC y aditivos y tampones de alta pureza. Si el problema persiste elimine uno a uno los componentes de la fase móvil hasta aislar el problema. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 152 - HPLC Troubleshooting and Maintenance El Reciclaje de fase móvil se encuentra limitado a las separaciones isocráticas exclusivamente, y es más útil para aquellos métodos empleados para análisis rutinarios con concentraciones de muestra moderadas como ocurre con frecuencia en los laboratorios de Control de Calidad. Los componentes de la muestra se diluyen en el tanque de recogida de fase móvil de forma que pasa un tiempo considerable hasta que el fondo (“background”) del cromatograma cambia de forma preciable. Esto se evidencia por lo general en una línea de base ascendente o un mayor nivel de ruido, produciéndose una pérdida en la sensibilidad de los analitos. En ese momento también pueden observarse picos fantasma de cuando en cuando. A menudo se añade ácido trifluoroacetico (TFA) a las fases móviles de fase reversa como agente formador de par-iónico para estrechar los picos cromatográficos. Potencialmente puede aparecer contaminación del TFA si se utiliza una única botella como origen, ya que la exposición del TFA al aire lo oxida rápidamente. Considere por lo tanto el uso de ampollas de TFA selladas individualmente más que alicuotar desde una botella ya abierta. Este puede ser especialmente aplicable para el análisis de estabilidad de muestra forzadas a degradarse. El Tetrahidrofurano sufre también de los mismos problemas de degradación oxidativa. La posibilidad de que el sistema de inyección pueda ser la fuente de picos fantasma también existe. Material proveniente de las muestras de inyecciones anteriores puede haber quedado adsorbido en diferentes componentes del sistema de inyección, especialmente en el rotor seal, y producirse unn “sangrado” en una escala grande tiempo. Evalúe lavando el sistema de inyección con un disolvente fuerte para inyectra posteriormente un blanco. Si no se observan picos en el blanco, optimice e incorpore ese régimen de lavado al método, o investigue el mecanismo potencial de la adsorción del componente y corríjalo en consonancia. Puesto que diferentes materiales de rotor pueden adsorber materiales diferentes de la muestra, merecería la pena investigar si el efecto se disminuye o incluso se elimina, cambiando entre rotores de Vespel, Tefzel y PEEK que están comercialmente disponibles. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 © Crawford Scientific 2007 - 153 - HPLC Troubleshooting and Maintenance 10.2.6 Picos Negativos La aparición de picos negativos se produce debido a factores no relacionados con las interacciones entre los analitos y la columna. Se observan picos Negativos cuando los cables de señal del detector están invertidos, o cuando el interruptor de polaridad se encuentra en la posición incorrecta. Los picos Negativos son normales cuando se utiliza el Detector de Indice de Refracción. Los picos Negativos aparecerán en el cromatograma si la absorbancia de la muestra es menor que la de la fase móvil a la longitud de onda de detección. Hay que emplear por lo tanto fases móviles que muestren una absorbancia UV/Vis baja, o cambiar la longitud de onda de detección del analito. Si a lo largo del tiempo se concentran en la fase móvil compuestos fuertemente absorbentes puede aparecer una situación similar si el disolvente se recicla. Si está empleando reciclado de fase móvil y comienzan a aparecer picos negativos tras un uso prolongado de la fase móvil, lave abundantemente la columna con un disolvente fuerte y re-equilíbrela, con fase móvil recientemente preparada. Si desaparece el problema de picos negativos, es un indicio de que la fase móvil estaba contaminada. Cuado se emplea el detector de Diodo Array (DAD) es recomendable emplear una longitud de onda de referencia para posibilitar la sustracción de línea de base. Esto tiene la ventaja de corregir una línea de base con deriva mejorando así los límites de detección y de cuantificación. Es importante asegurarse de que la longitud de onda de referencia y el ancho de banda (bandwith) se han elegido correctamente para prevenir la posibilidad de que se produzca una absorbancia mayor que la de los analitos a detectar y se generen picos negativos en el cromatograma. El procedimiento correcto para la selección de las longitudes de onda de detección y de referencia se describe en relación con el espectro UV del ácido anísico. Para optimizar la detección de la menor cantidad posible de Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 154 - HPLC Troubleshooting and Maintenance ácido anísico seleccione la longitud de onda de medida al pico más intenso del espectro, es decir, 252nm. Tratando el espectro como si fuera un pico pseudo-cromatográfico, compruebe su anchura a media altura (PWHH). Este será en ancho de banda de la longitud de onda de medida (Bandwith) p.ej. 30nm. Es posible reducir los efectos de fondo gracias a una adecuada elección de las longitudes de onda de medida y de referencia. La longitud de onda de referencia puede emplearse para eliminar los siguientes efectos, que provocarán todos ellos derivas de línea de base durante el análisis cromatográfico o bien mostrarán ondulaciones o ruido en la misma; 1. 2. Reducción del ruido generado por: • Particulas en suspensión • Cambios en el índice de refracción del eluyente cuando se emplean gradientes de elución. • Turbulencias en la celda de flujo Reducción en la deriva del instrumento causada por: • Envejecimiento / deformacion de la lámpara Identifique en el cromatograma el pico que absorbe a la longitud de onda mayor, en el ejemplo es el ácido anísico. Determine la longitud de onda a la que el pico del ácido anísico deja de absorber, se considera generalmente cuando la absorbancia cae por debajo de 1.0mAU, y en el ejemplo del ácido anísico será 300nm. Para evitar la posibilidad de una contribución a la absorbancia de la longitud de onda de referencia elegida, añada 10nm a este valor − Esta longitud de onda será el límite inferior de la longitud de onda de referencia, i.e. 310nm. Para eliminar los efectos de fondo descritos hay que elegir un ancho de banda grande, típicamente de 100nm, y situar la longitud de onda de referencia en el punto medio de esta ventana i.e. 360nm. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 © Crawford Scientific 2007 - 155 - HPLC Troubleshooting and Maintenance 10.3 Tiempo de Retención El tiempo de retención de los analitos puede fluctuar, aumentar o disminuir, y es crítico para diagnosticar y eliminar problemas en el sistema de HPLC. El diagnóstico de las variaciones observadas en el tiempo de retención equiere determinar si el problema está generado por el sistema HPLC o por la columna. A partir de los principios fundamentales; • • Si el tiempo muerto (t0) y el tiempo de retención del analito (tR) varían juntos podemos sospechar de un cambio en el flujo de eluyente. En este escenario el factor de capacidad (k') se mantendrá constante. Si es sólo el tiempo de retención del analito tR el que varía, entonces k' cambiará también. En este escenario las sospechas van dirigidas a un en cambio en la selectividad del sistema de separación, es decir, en la composición de la fase móvil, la temperatura, la fase estacionaria de la columna, etc. 10.3.1 Tiempos de Retención Fluctuantes La variación en el tiempo de retención de los analitos se debe generalmente a cambios producidos en la composición de la fase móvil, y es típicamente el resultado de una mezcla de solventes inconsistente. Una reducción del 5−10% en el tiempo de retención del analito por fase reversa puede producirse por cada 1% de incremento en la proporción del componente orgánico de la fase móvil. Esta variación es bien conocida, y a menudo se utiliza para producir grandes cambios en la selectividad particular de un sistema de separación para un rango de analitos cuando es necesario. La figura a continuación muestra la variación de tiempo de retención en una serie de inyecciones consecutivas de una mezcla estándar de cuatro componentes, empleando un sistema de mezcla a baja presión. La parte inicial del método de separación emplea una sección isocrática de 12 mn a 62% B. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 156 - HPLC Troubleshooting and Maintenance Suponiendo que el contenido de las botellas de disolvente se ha preparado correctamente, la incorrecta composición de la fase móvil puede provenir de unos de los dos problemas siguientes; 1. Si existe una restricción en uno o más de los canales de disolvente que vienen desde las botellas hacia la válvula de proporción, la proporción obtenida de la mezcla de solventes será incorrecta. Para eliminar la posibilidad de que el suministro de una o más fases móviles esté restringido, hay que desconectar la línea de entrada de disolvente de la válvula proporcional. Una vez desconectada, el líquido debe sifonar libremente si no existe ninguna restricción. Afloje el tapón de la botella de disolvente si está cerrado, para liberar cualquier vacío parcial de la botella. Si la presurización insuficiente fuese la causa del problema el solventes sifonará entonces perfectamente. Si el flujo queda aún restringido, quitar el filtro de entrada de disolvente y comprobar si sifona de nuevo. Si la línea sifona libremente el filtro está bloqueado. Si el filtro es del tipo de vidrio sinterizado límpielo sumergiéndolo en ácido nítrico 6M, para después lavarlo abundamentemente con H2O, después con MeOH, y de nuevo con agua antes de reinstalarlo. No lo sonique, puesto que podría provocarse la fractura del vidrio debido a la frecuencia de ultrasonidos aplicada. Si una línea de disolvente se encuentra restringida se introducirá menos disolvente, generando un ligero vacío en la válvula proporcional de gradiente. En consecuencia, cuando la segunda válvula se abra habrá un torrente del nuevo disolvente para compensar ese vacío parcial, con el resultado de que la proporción de disolvente introducido variará. Un exceso de disolvente B en la fase móvil hará que los analitos eluyan antes, como en el ejemplo observado en el cromatograma del ejemplo. 2. Si el suministro de disolvente no se encuentra restringido, el problema puede encontrarse en el software de control o sospecharse de un problema mecánico en la válvula proporcional. Los sistemas de mezcla a baja presión trabajan abriendo cada válvula proporcional durante un tiempo determinado, cerrándola, y abriendo después la segunda válvula, etc. En el ejemplo anterior, si el ciclo total de la válvula fuese de 100ms, y se necesitara una fase móvil isocrática de 38% A:62% B la válvula A permanecerá abierta durante 38ms y la válvula B durante 62ms. Para comprobar si hay fallos en la válvula proporcional, cebe todas las líneas con el mismo disolvente y partiendo de volúmenes iguales en las botellas realice una mezcla cuaternaria de proporción fija 1:1:1:1 (v/v) durante un tiempo fijo a un flujo fijo. La reducción del volumen en cada botella de disolvente debe ser la misma si la válvula funciona correctamente. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 © Crawford Scientific 2007 - 157 - HPLC Troubleshooting and Maintenance Adicionalmente, cuando se dejan preparadas fase móviles durante un tiempo los solventes más volátiles de la mezcla pueden evaporarse, produciendo un cambio en las características generales de retención. Sin embargo la evaporación no tiene mucha influencia en los métodos en fase reversa, a menos que la botella de disolvente se caliente durante el uso. Pueden producirse problemas con la fase móvil cuando se usa un disolvente reciclado incorrectamente. Asegúrese de que la botella de disolvente contiene un minímimo de unos tres litros de fase móvil, y que está siendo agitada constantemente. De esta forma los contaminantes o cualquier otro pequeño cambio en el eluyente proveniente de la columna serán diluidos eficientemente antes de pasar a través del sistema la próxima vez. Las variaciones de tiempo de retención también pueden ser causadas por fluctuaciones de la temperatura. Por cada cambio de 1ºC en la temperatura de la columna puede producirse un cambio del 1−2% en el tiempo de retención. La forma más sencilla de eliminar este problema potencial es asegurarse de que tanto la columna como la fase móvil están termostáticamente controladas. Compruebe problemas potenciales de temperatura usando un termómetro calibrado, y determine si cualquier fluctuación de temperatura observada se correlaciona con cambios en el tiempo de retención de los analitos. El envejecimiento de la columna puede también contribuir a variaciones en el tiempo de retención. El empleo de guarda columna puede extender en la práctica la vida efectiva de una columna. Más aún, se recomienda el uso de guarda columna por una razón específica; servir de filtro para eliminar materiales fuertemente retenidos que podrían contaminar potencialmente la columna analítica. Analizar estándares de comprobación con más frecuencia puede permitir un sistema de captura de datos que actualice convenientemente la deriva observada en el tiempo de retención. Asigne un pico cromatográfico de la muestra como pico de referencia. El uso de un estándar interno puede ayudar también, en especial si se utilizan tiempos de retención relativos en lugar de absolutos. La tabla siguiente muestra el efecto observado de cambiar las condiciones de separación en el tiempo de retención tR de los analitos. Efecto del cambio de las Condiciones de Separación en la retención de los analitos Variable Método Cambio Variable Cambio Prom. tR Flujo Todos +1% -1% Temperatura Todos salvo SEC +10C -(1−2%) Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 158 - HPLC Troubleshooting and Maintenance Composición de la Fase Móvil: Solvente Orgánico RP-HPLC* +1% (v/v) -(5−10%) pH RP-HPLC +0.01 unit ±(0−1%) Solvente Fuerte Fase Normal +1% -(1−2%) Tampón: SEC +1% 0% Solvente Orgánico * Incluyendo Cromatografía de Pares de Iones RP-HPLC Mejora la fiabilidad de cualquier análisis LC con respecto a las variaciones en tiempo de retención y a la forma de los picos asegurándose de que la capacidad tamponadora de la fase móvil es suficiente para compensar cualquier cambio de pH. Generalmente un tampón funcionará con la mayor eficiencia cuando se encuentre en ±1 unidades de pH del pka del analito. Es muy importante saber que los tampones fosfato no proporcionan la capacidad tamponadora deseada en el rango de pH de 3−6. Este rango de pH puede llenarse bien usando un tampón acetato (pka 4.8), o citrato, ya que este tampón muestra tres pkas en el rango de pH típico de las separaciones en fase reversa. Sin embargo, el citrato tiene el inconveniente de ser altamente corrosivo para las superficies de acero inoxidable. La tabla siguiente muestra el rango efectivo de la capacidad tamponadora de diferentes tampones ácidos comunes: Valores pka para Tampones Ácidos comunes Tampón Pka Rango pH efectivo* Fosfato 2.1 1.1 − 3.1 7.2 6.2 − 8.2 12.3 11.3 − 13.3 Acetato 4.8 3.8 − 5.8 Citrato 3.1 2.1 − 4.1 4.7 3.7 − 5.7 5.4 4.4 − 6.4 * Rango pH efectivo − ±1 pH unidades del pka Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 © Crawford Scientific 2007 - 159 - HPLC Troubleshooting and Maintenance Para mantener una adecuada capacidad tamponadora de las muestras de analitos, comience con una concentración de tampón entre 25−50mM. No emplee menos de 20mM, a no ser que utilice un detector espectrométrico de masas. Considere cambiar a un sistema tampón de naturaleza volátil p.ej. acetato amónico o formiato amónico. Los tampones volátiles previenen contra la contaminación y el ensuciamiento de la fuente del espectrómetro de masas, mejorando la sensibilidad y la eficiencia en la detección. La figura de abajo muestra el efecto negativo producido por variación del pH en el tiempo de retención del analito y en su forma de pico. Los dos compuestos cromatografiados son ácido benzoico y sórbico respectivamente. A un pH tamponado de 3.5 éstos ácidos débiles se encuentran completamente protonados, y por lo tanto muestran tiempos de retención largos en una caolumna de fase reversa. (Cromatograma A). A un pH tamponado a pH 7.0 los ácidos se encuentran completamente desprotonados. Poseen ahora un grado de polaridad mucho mayor que a pH 3.5, y en consecuencia su tiempo de retención decrece dramáticamente (Cromatograma B). Sin embargo, se utiliza un pH no tamponado de 7.0 el estado de ionización de estos analitos no está controlado de forma eficiente, y el equilibrio dinámico está constantemente cambiando entre la forma ionizada e ion- suprimida y tanto el tiempo de retención como la forma de pico varían dramáticamente (Cromatograma C). 10.3.2 Decrecimiento del Tiempo de Retención Si el tiempo muerto(t0) y el tiempo de retención (tR) decrecen simultáneamente, el factor de capacidad del analito (k') permanecerá constante. En este escenario puede sospecharse de un progresivo aumento en el flujo. Compruebe los parámetros de operación del método y la calibración del flujo del sistema. Chequee el pH de cualquier fase móvil tampón utilizada. A menos que se establezca específicamente por parte del fabricante de la columna, el pH de operación debe mantenerse en el rango de pH de 2 a 8. Por debajo de pH 2 el enlace siloxano que une el ligando de la fase estacionaria al soporte de sílice se romperá, produciéndose la pérdida de fase estacionaria. Por encima de aproximadamente pH 8 se produce la disolución del soporte de sílice. En ambos casos disminuirá el tiempo de retención de los analitos. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 160 - HPLC Troubleshooting and Maintenance Considere cambiar el tipo de columna a una variedad polimérica no basada en sílice para permitir separaciones cromatográficas a pH extremo. Asegúrese de que la columna no esta sobrecargada. Aplique la ecuación descrita en la pág 145, y si fuera necesario aumente la carga de carbono de la columna utilizada, o disminuya la cantidad absoluta de la masa de analito aplicado bien diluyendo la muestrta o reduciendo el volumen de inyección. Elimine cualquier interacción secundaria de la columna usando un modificador de fase móvil o tamponando ésta adecuadamente. La Trietilamina puede añadirse como base competitiva para eliminar las interacciones polares con los grupos silanol residuales, y para aumentar la carga de carbono efectiva de la columna. Si realiza gradientes de elución asegúrese de que todos componentes del disolvente se mezclan en la proporción y calidad de mezcla adecuadas. Afloje los tapones de las botellas de disolvente para liberar cualquier formación de vacío parcial, asegurando la completa presurización y el llenado completo de las líneas de alimentación de disolvente. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 © Crawford Scientific 2007 - 161 - HPLC Troubleshooting and Maintenance 10.3.3 Aumento del Tiempo de Retención Si el tiempo muerto(t0) y el tiempo de retención (tR) aumentan simultáneamente, el factor de capacidad del analito (k') permanecerá constante. En este escenario puede sospecharse de una progresiva disminución en el flujo. Compruebe los parámetros de operación del método y la calibración del flujo del sistema. Compruebe el cabezal de la bomba buscando fugas. Si realiza gradiente de elución compruebe que el gradiente está siendo mezclado y proporcionado correctamente. Afloje los tapones de las botellas de disolvente para liberar cualquier formación de vacío parcial, asegurando la completa presurización y el llenado completo de las líneas de alimentación de disolvente. 10.4 Incidencias con la Presión Durante el funcionamiento puede esperarse algún aumento en la contrapresión del sistema. Un aumento en la viscosidad de la fase móvil producirá un incremento en la presión del sistema, y puede producirse simplemente por una reducción de la temperatura de operación de la columna. De forma similar cualquier cambio en la composición de la fase móvil puede alterar la viscosidad. El Isopropanol es cuatro veces más viscoso que el metanol a temperatura ambiente, mientras que una mezcla de H2O:MeOH 40:60 (v/v) generará aproximadamente el doble de la presión que el MeOH puro. Otros factores que contribuirán a un incremento en la presión del sistema son el aumento del flujo, el aumento de la longitud de la columna, la reducción del tamaño de las partículas de sílice o cualquier bloqueo o restricción en la ruta seguida por la fase móvil. Elimine la columna como causa del aumento de presión retirándola del sistema y sustituyéndola por una unión de volumen cero. Para localizar un bloqueo parcial reduzca el flujo de fase móvil de forma que se obtenga una presión inferior a 500psi. Con las gafas de protección puestas, vaya aflojando sistemáticamente las conexiones LC, yendo desde el detector hacia la bomba. Cuando se afloje la conexión justo antes del componente bloqueado la presión caerá de forma significativa. La pieza sospechosa puede limpiarse o sustituirse si es necesario. Si después de eliminar el bloqueo éste vuelve a aparecer al poco tiempo, debemos examinar con mayor detalle la fuente potencial del problema. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 162 - HPLC Troubleshooting and Maintenance 10.4.1 Aumento Continuo de la Presión sin Inyecciones de Muestra Cuando se observa un aumento constante en la presión del sistema sin que se hayan realizado inyecciones las causas pueden ser las siguientes: 1. Partículas en la Fase Móvil Deben filtrarse todos los disolventes y tampones que no sean de gradoHPLC a través de un filtro de 0.22µm para eliminar cualquier material particulado; ver págs 40 ss para más información. Las partículas en la fase móvil pueden dañar los sellos de los pistones del cabezal de la bomba. El desgaste de los sellos de los pistones puede provocar que las válvulas antirretorno se peguen produciendo incrementos de presión, además de las variaciones en el tiempo de retención de los analitos debido al inadecuado suministro de fase móvil. Cuando se sustituyan los sellos es necesario evaluar también el desgaste de los pistones. Con una lupa y luz intensa compruebe que no se han formado estrías o canales en el émbolo del pistón. Cualquier daño del pistón producirá inmediatamente un daño en el sello reproduciendo el problema. 2. Los componentes de la Fase Móvil son inmiscibles o insolubles. Compruebe la compatibilidad de los disolventes de la fase móvil usando la tabla de compatibilidad de la pág 15. Emplee un disolvente intermedio de lavado cuando tenga que cambiar entre fases móviles potencialmente inmiscibles. Elimine cualquier traza de tampón con agua antes de la siguiente aplicación, para evitar la posible precipitación y la introducción inadvertida de material particulado en el sistema cuando se cambia de un método a otro. 3. Fase Móvil Inestable (polimerización). 4. Formación de volúmenes vacíos en la columna Las prestaciones de la columna se irán deteriorando con el tiempo. La exposición continua a alta presión producirá la compresión de la fase estacionaria, y el desarrollo de un volumen vacío en cabeza de columna. Además del aumento en la contrapresión, la forma del pico cromatográfico comenzará a deteriorarse; se producirá ensanchamiento de los picos debido a la caída de la Eficiencia y aparecerán desdoblamientos de los picos y hombros. La disolución de la sílice se produce a pH alto. A menos que lo establezca específicamente el fabricante, no use tampones con pH por encima de pH 8. Al trabajar con una fase móvil a pH alto debe emplearse una columna saturadora que puede ayudar a prolongar la vida de la columna analítica. Una columna saturadora puede ser una columna vieja del mismo tipo que la columna analítica situada entre el cabezal de la bomba y el automuestreador. Para minimizar la Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 © Crawford Scientific 2007 - 163 - HPLC Troubleshooting and Maintenance contrapresión adicional producida la columna saturadora suele ser de 150 mm o más corta. El fundamento técnico es que la fase móvil iniciará el ataque a la columna saturadora en primer lugar, pre-tratando de forma efectiva la fase móvil de forma que será menos agresiva con la columna analítica. A medida que el relleno de la columna saturadora se va disolviendo se generan micropartículas de sílice que van acumulándose y bloqueando la frita de salida de la columna saturadora. Estas partículas se van haciendo cada vez más pequeñas, y eventualmente pueden pasar a través del filtro de salida de la columna saturadora. Es muy conveniente por lo tanto instalar un filtro de línea de 0.5µm o menor entre la salida de la columna saturadora y la entrada del automuestreador para atrapar estas partículas. Con la disponibilidad comercial de rellenos de sílice estables a alto pH, y de fases sin sílice resistentes al ataque pH, actualmente hay menos justificación en el uso de columnas saturadoras. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 164 - HPLC Troubleshooting and Maintenance 10.4.2 Aumento Continuo de la Presión con Inyecciones de Muestra 1. Particulas en la muestra Las partículas existentes en la muestra producen contaminación en la frita de entrada de la columna, generando un incremento de la presión y anormalidades en la forma de los picos p.ej. colas, desdoblamientos, etc. Revise visualmente la calidad de la muestra disuelta antes de inyectarla para decidir si necesita o no ser filtrada. Si el aspecto es turbio o hay visible material particulado, filtre a través de un filtro de 0.5 o 0.22µm. Instale un filtro de línea entre el inyector y la columna, y si se sospecha de contaminación proveniente de la muestre instale una guarda columna para servir de filtro químico y de partículas. 2. Compuestos de la Muestra insolubles en la Fase Móvil. Bajo condiciones de inyección ideales el disolvente de la muestra debe ser el mismo que la fase móvil, o más débil. De esta forma se evitarán anormalidades en la forma de los picos ya que el disolvente de la muestra no requiere dilución a la concentración de la fase móvil y se evitan mecanismos de partición secundarios. Refiérase a las págs 79−82 para más información sobre las condiciones a aplicar cuando se inyecten muestras en solventes de mayor fuerza eluotrópica que la fase móvil para asegurar la solubilidad. 3. Componentes de la muestra irreversiblemente enlazados a la fase estacionaria de la columna. Asegúrese de que todos los componentes de la muestra son eluidos de la columna después de su uso. Elimine los analitos fuertemente adsorbidos lavando la columna con un disolvente de gran poder de elución. Asegúrese de que el poro de la fase estacionaria es suficiente para analizar los analitos en cuestión; use columnas con tamaño de poro de 300Å para compuestos de peso molecular mayor de 5kDa. Invierta el flujo de fase móvil a través de la columna para acelerar la eliminación de cualquier analito fuertemente retenido o liberar el bloqueo parcial del filtro de entrada de la columna. Las columnas con base de sílice pueden invertirse generalmente sin consecuencias. Sin embargo, algunas columnas especiales como las de Filtración en Gel o las de resinas de intercambio iónico pueden no ser tan robustas. Consulte la literatura proporcionada por el fabricante de la columna para aclarlo. Desconecte la columna, inviértala y conecte la antigua salida al tubo que viene del automuestreador. Lave aproximadamente con 20 volúmenes de columna de fase móvil ~ 50mL para una columna de 4.6 x 250mm para eliminar cualquier partícula de la frita, antes de Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 © Crawford Scientific 2007 - 165 - HPLC Troubleshooting and Maintenance conectar su salida (la antigua entrada) al detector. Deje la columna en esta configuración de flujo inverso y continúe la operación normalmente. 10.5 Sensibilidad Una baja sensibilidad puede producirse por alguna de las razones siguientes: 1. • • 2. • • • • 3. • • • 4. • • ¿Hay suficiente muestra? Compruebe pérdidas durante la preparación de la muestra realizando análisis de muestras dopadas con una concentración conocida de analito. Considere el uso de estándar interno para ajustar las pérdidas durante la el proceso de preparación de muestra optimizando el método de forma apropiada. ¿Problemas en el Inyector? Compruebe la calibración del automuestreador. Aplique la técnica de inyección adecuada para el volumen disponible de muestra. Consulte las págs 79-82 para más información sobre el llenado completo o parcial del loop. La aguja del Inyector está parcialmente obstruida. Rotor Seal desgastado, con una ruta no barrida por la fase móvil. ¿Problemas en el Detector? Compruebe el flujo de fase móvil a través del detector, despejando cualquier posible bloqueo. La atenuación del Detector es demasiado alta. Es importante verificar los datos cromatográficos proporcionados por el sistema de tratamiento de datos periódicamente, evaluando el aspecto de la línea de base, picos y separación en general. Con el fin de evaluar adecuadamente la sensibilidad del sistema es necesario disponer se una vista estándar o atenuación del instrumento. Aunque la atenuación puede fijarse en el propio instrumento, es más frecuente que la realice el sistema de tratamiento de datos. Un usuario puede pensar que tiene mala sensibilidad frente a un analito si la atenuación se ha seleccionado muy alta. Concentración de la Muestra fuera del rango lineal del detector. Diluya o pre-concentre la muestra para llevar a los analitos dentro del rango lineal del detector. ¿Cambio en el Flujo? Flujo Reducido Un flujo reducido generará picos cromatográficos anchos.Las áreas de pico se mantendrán constantes, aunque la altura de pico se reducirá, reduciendo la sensibilidad efectiva y los límites de detección y cuantificación. Compruebe los ajustes del método. Evalúe el sistema de suministro de disolvente para comprobar si los sellos de los pistones se han desgastado o las válvulas antirretorno se han pegado o bloqueado parcialmente afectando al fujo de fase móvil. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 - 166 - HPLC Troubleshooting and Maintenance • La reducción de la temperatura de separación, la viscosidad de la fase móvil, cambian el flujo efectivo y los efectos de transferencia de masa. 10.6 Recuperación Una mala recuperación de la muestra puede producirse por múltiples razones, pero se debe típicamente a la adsorción irreversible de los ananlitos en la fase estacionaria o en los componentes extra-columna. Aumente la fuerza eluotrópica de la fase móvil para minimizar la adsorción. Para la separación de analitos básicos emplee sílice desactivada, columnas con en-capping, o añada una base competitiva como la trietilamina para minimizar las interacciones retentivas secundarias. Considere usar una columna de fase estacionaria pomérica si la adsorción en demasiado fuerte. Emplee componentes inertes en las conexiones y tubos. Traducción Grupo Biomaster 2009 © Crawford Scientific 2007 © Crawford Scientific 2007 - 167 -