arterosclerosis 120609114038 phpapp02
Anuncio

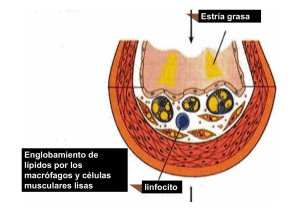
Universidad nacional José Faustino Sánchez Carrión ES C U E L A P RO F ES I O N A L D E ENFERMERIA Integrantes: Bernal Paulet Antony Ramírez Leandro Víctor Arterosclerosis • Enfermedad localizada de las arterias, caracterizada por la existencia de placas que protuyen en la luz vascular, compuestas de lípidos, células de músculo liso, tejido conectivo amorfo y restos celulares; que en su evolución puede dar lugar a la aparición de déficits isquémicos en los territorios irrigados por los vasos donde asienten estas lesiones. Proceso de la arterosclerosis Normal estría grasa Placa Fibrosa Placa Aterosc lerotica Placa de ruptura / fisura y trombosis Clinicamente silencioso Claudicación intermitente angina estable Angina Inestable IM }ACS Accidente cerebrovasc ular isquémico / TIA Isquemia crítica de la pierna muerte cardiovascular Incremento de edad ACS, el síndrome coronario agudo, ataque isquémico transitorio, accidente isquémico transitorio 1) Interacciones celulares y moleculares • La acumulación de LDL (lípidos de baja densidad) OXIDADO en la capa íntima (tejido conjuntivo denso) contribuye al reclutamiento de monocitos y a la formación de células espumosas. • Inicio de la lesión • El endotelio vascular es una barrera selectivamente permeable entre sangre y tejidos. • Genera moléculas efectoras que regulan la formación de trombos, el tono vascular, la inflamación y remodelación arterial. • Al remover el endotelio, las células musculares lisas migran hacia el lumen y proliferan hasta que se restablece el endotelio vascular. 2) Acumulación DE LDL en la matriz subendotelial. • • • • • • Como es lógico, la retención de LDL es mayor cuando los niveles circulantes son altos. El atrapamiento del LDL se produce entre sus apo B lipoproteínas y los proteoglicanos de la matriz. Las apo B lipoproteínas son segregadas por el hígado como VLDL (very low density lipoprotein). Se convierte en LDL cuando es removido el triglicerol y la apolipoproteína C, del VLDL (probablemente en el plasma e hígado). Lipoproteínas sólo pueden acumularse en la capa íntima. Contienen un polipéptido adicional que forma un puente disulfuro con proteínas arteriales. Los glóbulos de LDL atrapados, sufren OXIDACIÓN, LIPOLISIS, PROTEOLISIS y AGREGACIÓN. Estos cambios aumentan la inflamación y la formación de células espumosas que tienden a ser fagocitadas por macrófagos. 3) Oxidación De Lípidos En La Pared Vascular • Las enzimas Lipooxigenasa insertan oxigeno molecular en ácidos grasos, formando lipoperóxidos, que son fácilmente transferidos a través de la pared vascular y se unen al LDL. • HDL • Las lipoproteínas de alta densidad poseen una FUERTE ACTIVIDAD ANTIARTEROESCLEROTICA, pues remueve el exceso de colesterol de los tejidos y PROTEJE A LAS LIPOPROTEINAS DE SU OXIDACION. • Su actividad antioxidante (del HDL) se debe a la PAROXENASA SERICA, una enzima estearasa, que se lleva sobre el HDL. Puede degradar ciertos lípidos oxidados. LESION INICIAL ESTRÍA GRASA PLACA ATEROMA Acumulación de LDL oxidadas Aumento de células musculares lisas y colágeno FIBROATEROMA Fibrosis/ruptura/trombosis La formación de la placa aterosclerótica comienza desde la más temprana adolescencia 4) Inflamación • Se produce con el reclutamiento de linfocitos y monocitos por la pared • • • • • arterial. El disparador es la acumulación mínima de LDL OXIDADO en el endotelio, el cual es estimulado a generar moléculas pro inflamatorias como las moléculas de adhesión y factores de crecimiento (factor estimulador de macrófagos). La actividad biológica del LDL oxidado está principalmente en la fracción fosfolípida del LDL (tres productos activos se han identificado, derivados de ACIDOS GRASOS INSATURADOS –peróxidos de lípidos) EL OXIDO NITROSO es un mediador celular con múltiples propiedades antiaterogénicas, incluyendo la VASODILATACIÓN, que disminuye la presión arterial y dificulta la detención de trombos. Ratones que carecen del gen que produce la enzima óxido nítrico sintetasa poseen ateroesclerosis más desarrollada y presión sanguínea alta (Ratones knock-out). Una CITOQUINA (M-CSF), producida por el endotelio, estimula la diferenciación y proliferación de macrófagos. 5) Formación De Células Espumosas • Los lípidos deben ser oxidados en las partículas de LDL para que puedan ser tomados (fagocitadas) por los macrófagos. • En la oxidación están implicados radicales oxidantes producidos por el endotelio (respiración aerobia en la mitocondria), enzimas plasmáticas o segregadas por los tejidos arteriales como: MIELOPEROXIDAS, ESFINGOMIELINASA, FOSFOLIPASAS, etc. • MIELOPEROXIDAS: produce radicales libres altamente oxidantes como el ácido hipocloroso (HClO), el radical tirosilo, y modifica al LDL para unirse a receptores en los macrófagos. • ESFINGOMIELINASA: promueve agregación de lipoproteínas que se retienen en la pared arterial y facilita la fagocitosis de macrófagos ("engordan" más de grasas). • FOSFOLIPASA (grupo II sPLA") promueve la oxidación del LDL. También es ratones transgénicos (knock-out), sin el gen de esta enzima desarrollan rápidamente la dolencia. PLACAS FIBROSAS • La forman LIPIDOS EXTRACELULARES, COLESTEROL esterificado con ácidos grasos y CELULAS MUSCULARES LISAS. Factores que favorecen la formación de placas fibrosas: • HOMOCISTEINA, lesiona al endotelio y estimula la proliferación de células musculares • HIPERTENSIÓN • Algunas hormonas • La angiotensina II que eleva la presión arterial, también estimula la proliferación de células musculares lisas en la pared arterial. Existen medicamentos que disminuyen la producción de angiotensina, inhibiendo la enzima "angiotensinasa", y bajando la presión arterial. • EL ORIGEN MONOCLONAL DE CELULAS MUSCULARES LISAS EN LAS LESIONES ATEROESCLEROTICAS, puede envolver una transformación no maligna de las mismas (monoclonal significa que se reproducen en la lesión a partir de una sola célula). Formación de la placa también puede causar el endurecimiento de las arterias, resultando en debilitamiento y adelgazamiento de la pared del vaso, dando lugar a un aneurisma y posiblemente hemorragia. Adelgazamiento de la cápsula fibrosa Hemorragia desde microvasos de la placa Ruptura de la cápsula fibrosa Daño endotelial Respuesta protectora lleva a la producción de moléculas de adhesión celular Monocitos y linfocitos T adhieren a La superficie ‘pegajosa’ de las células endoteliales Migración hacia espacio subendotelial Macrófagos atrapan LDL-colesterol oxidado Células espumosas ricas en lípidos Estría lipídica y placa aterosclerótica Síndrome Clínico Inestable de Aterosclerosis El cardíaco El mesentérico El cerebral Dependiendo del territorio isquémico puede tener unas u otras repercusiones siendo algunos órganos o vísceras más sensibles . Territorios como: Manifestación Clínicas Aterosclerosis Coronaria Infarto del miocardio Angina de pecho Muerte súbita de causa cardiaca Existen tres formas clásicas de las presentación clínica de la aterosclerosis coronaria Primera forma de presentación clínica de arterosclerosis coronaria Mujeres síntomas menos frecuencia. Dolor torácico característico de isquemia del miocardio Angina de Pecho Síntomas de angina estable son sutiles y difíciles de distinguir Retro esternal con irradiación al brazo izquierdo y al cuello Disnea Angina de pecho • Es un concepto exclusivamente clínico y su diagnostico se basa en las características y circunstancias que acompañan en el dolor • Los mecanismos que lo provocan: Aumento por la necesidad de oxigeno provocadas por : • Cambios de la presión arterial ejercicio • Cambios en la frecuencia cardiaca emociones Angina de pecho • Debido al un déficit de oxigeno , la glucosa produce energía por medio de la Glucolisis Anaerobia , con la producción de cristales de piruvatos y lactatos, con la consiguiente ACIDOSIS METABÓLICA • Acidosis láctica: proceso caracterizado por la acumulación de ácido láctico en la sangre, provocando la disminución del pH en el músculo y en el plasma. Segunda forma de presentación clínica de aterosclerosis coronaria. Infarto del Miocardio Si no se trata la angina IM Dolor torácico no obstante, puede ser más persistente y más intenso Síntomas autónomos como nauseas y vómitos. Pueden aparecer arritmias En casos más graves, puede haber síntomas de insuficiencia cardiaca. Infarto de miocardio • • • • Si el trombo producido obstruye casi el 90% de la pared del vaso el flujo del vaso se vera seriamente puede causar deficiencia del aporte de oxigeno El metabolismo normal del miocardio es aeróbico (ATP obtenido a través de la fosforilacion oxidativa) Se reduce el flujo de sustratos hacia el área afectada y también le eliminación de los mismos lo que aumenta la presión oncotica intracelular produciendo tumefacción celular lo que afecta la permeabilidad de la membrana plasmática Esto produce un agotamiento de ATP , acumulación de acido láctico, aparición de acidosis grave y notoria reducción de la fuerza contráctil Aparición de aterosclerosis y rotura de la placa Formación de un trombo grande en una arteria coronaria – produciendo isquemia Desviación hacia la glucolisis (anaeróbica ) síntesis disminuida de ATP Acumulación de acido láctico y otros meta bolitos en el musculo miocárdico , aumentando la alteración de la permeabilidad de la membrana Disminución del Ph y contracción ineficiente Cese de la contracción y activación de fosfolipasas de membrana, degradación de proteínas por proteasas Muerte del área del musculo afectada Tercera forma de presentación de aterosclerosis coronaria Muerte Súbita de causa Cardiaca Es la primera manifestación clínica en 25% de las personas que presentan la enfermedad. Única esperanza para MSC es la Reanimación cardiopulmonar y desfibrilación ventricular. La reanimación tras la MSC es más eficaz en las personas ingresadas al hospital. Muerte súbita • Ocurre minutos después de que aparecen los síntomas. La causa subyacente mas común del fallecimiento súbito por paro cardiaco en pacientes es la enfermedad coronaria (acumulación de grasa en las arterias que suministran sangre al musculo coronario) • Esto produce una deficiencia del latido cardiaco que lleva a un paro cardiaco